


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Nakaniwa, Tetsuko*; Fukata, Harumi*; Inoue, Tatsuya*; Goda, Masaki*; Nakai, Ryoko*; Kirii, Yasuyuki*; Adachi, Motoyasu; Tamada, Taro; Segawa, Shinichi*; Kuroki, Ryota; et al.
Biochemistry, 51(42), p.8410 - 8421, 2012/10
Times Cited Count:18 Percentile:42.84(Biochemistry & Molecular Biology)Protein kinase is a vital drug target for the treatment of a wide range of diseases. To investigate the effect of cysteine mutation on the function, stability and structure of kinase, free cysteines of c-Jun N-terminal kinase 1 (JNK1) were systematically removed by mutation. Two cysteine-destructed mutants in which three (M3) and seven (M7) cysteine residues are removed, yielded about 5 and 2 times than wild type JNK-1 (M0). SDS PAGE analysis showed that the aggregation was less in the case of M3 and M7. Thermal unfolding experiment of M0, M3 and M7 using by differential scanning calorimetry proceeded at least three state unfolding. Crystal structure of the M3 mutant was determined to 2.6 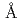 resolution, which was identical to that of the wild-type. Consequently, due to the highest yield, its improved stability against aggregation and its structural similarity to the wild type, the M3 mutant is suitable for the use of further characterization of its function and structure.
resolution, which was identical to that of the wild-type. Consequently, due to the highest yield, its improved stability against aggregation and its structural similarity to the wild type, the M3 mutant is suitable for the use of further characterization of its function and structure.
Yamasaki, Chisato*; Murakami, Katsuhiko*; Fujii, Yasuyuki*; Sato, Yoshiharu*; Harada, Erimi*; Takeda, Junichi*; Taniya, Takayuki*; Sakate, Ryuichi*; Kikugawa, Shingo*; Shimada, Makoto*; et al.
Nucleic Acids Research, 36(Database), p.D793 - D799, 2008/01
Times Cited Count:52 Percentile:71.15(Biochemistry & Molecular Biology)Here we report the new features and improvements in our latest release of the H-Invitational Database, a comprehensive annotation resource for human genes and transcripts. H-InvDB, originally developed as an integrated database of the human transcriptome based on extensive annotation of large sets of fulllength cDNA (FLcDNA) clones, now provides annotation for 120 558 human mRNAs extracted from the International Nucleotide Sequence Databases (INSD), in addition to 54 978 human FLcDNAs, in the latest release H-InvDB. We mapped those human transcripts onto the human genome sequences (NCBI build 36.1) and determined 34 699 human gene clusters, which could define 34 057 protein-coding and 642 non-protein-coding loci; 858 transcribed loci overlapped with predicted pseudogenes.
