


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Suzuki, Kimichi*; Tachikawa, Masanori*; Shiga, Motoyuki
Journal of Chemical Physics, 138(18), p.184307_1 - 184307_7, 2013/05
Times Cited Count:10 Percentile:35.54(Chemistry, Physical)Temperature dependence on the structural fluctuations of Zundel cation, H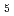 O
O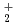 , and its isotopomers, DO and TO, have been studied using path integral molecular dynamics simulations in which nuclear quantum effect is fully taken into account. It has been found that the fluctuations of hydrogen-oxygen and oxygen-oxygen distances, which are relevant to the hydrogen bonded structure, grow drastically as the temperature increases within the range of investigation between 100 K and 900 K. The fluctuation with respect to the position of non-bonded hydrogen also increases substantially as the temperature increases. The temperature dependence on the fluctuation is greater for DO or TO than that of HO, since the zero-point effect of the former is less than the latter.
, and its isotopomers, DO and TO, have been studied using path integral molecular dynamics simulations in which nuclear quantum effect is fully taken into account. It has been found that the fluctuations of hydrogen-oxygen and oxygen-oxygen distances, which are relevant to the hydrogen bonded structure, grow drastically as the temperature increases within the range of investigation between 100 K and 900 K. The fluctuation with respect to the position of non-bonded hydrogen also increases substantially as the temperature increases. The temperature dependence on the fluctuation is greater for DO or TO than that of HO, since the zero-point effect of the former is less than the latter.
 H
H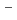 and FH
and FH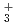
Suzuki, Kimichi*; Ishibashi, Hiroaki*; Yagi, Kiyoshi*; Shiga, Motoyuki; Tachikawa, Masanori*
Progress in Theoretical Chemistry and Physics, 26, p.207 - 216, 2012/08
The quantum nature of the strong hydrogen bonds for the FH and FH ions and their deuterated isotopomers at the room temperature has been studied using ab initio path integral molecular dynamics (PIMD) simulations. It is found that, for both of these ions, the hydrogen-bonded H/D atoms largely fluctuate around the central position of two F atoms. The average FH/FF distances of FH and FH are longer than the average FD/FF distances of F D
D and FH due to the primary/secondary isotope effects, which stem from the difference of the quantum nature of H and D nuclei. These results are compared with the family of Zundel-type ions, OH
and FH due to the primary/secondary isotope effects, which stem from the difference of the quantum nature of H and D nuclei. These results are compared with the family of Zundel-type ions, OH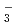 , NH
, NH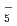 , OH
, OH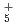 , and NH
, and NH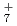 , which have been studied previously with the same ab initio PIMD approach. A comparison is also made with the previous experimental and ab initio vibrational configuration interaction results of FH.
, which have been studied previously with the same ab initio PIMD approach. A comparison is also made with the previous experimental and ab initio vibrational configuration interaction results of FH.
O)Koizumi, Akihito*; Suzuki, Kimichi*; Shiga, Motoyuki; Tachikawa, Masanori*
International Journal of Quantum Chemistry, 112(1), p.136 - 139, 2012/01
Times Cited Count:0 Percentile:0.05(Chemistry, Physical)Ab initio path integral molecular dynamics simulation of MOH(HO) (M= Cu, Ag, and Au) clusters has been carried out to analyze how the hydrogen-bonded proton can be affected by the counter noble metal cation. The CuOH(HO) cluster forms no hydrogen bonded structure even for the static equilibrium structures. Contrary to our previous paper of hydrated alkali metal hydroxide clusters, proton transferred distribution was not observed because of the high potential barrier heights of MOH(HO).
Koizumi, Akihito*; Suzuki, Kimichi*; Shiga, Motoyuki; Tachikawa, Masanori*
Journal of Chemical Physics, 134(3), p.031101_1 - 031101_3, 2011/01
Times Cited Count:14 Percentile:44.6(Chemistry, Physical)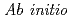 path integral molecular dynamics simulation of M
path integral molecular dynamics simulation of M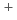 H
H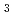 O
O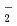 (M = Li, Na, and K) has been carried out to study how the structure and dynamics of low-barrier hydrogen-bonded Zundel anion, HO, can be affected by the counter alkali metal cation, M. Our simulation predicts that the quantum proton transfer in Zundel anion can be strongly coupled to the motion of counter cation located nearby. A smaller cation can induce larger structural distortion of the Zundel anion fragment making the proton transfer barrier higher, and hence lowers the vibration excitation energy. It is also argued that a large H/D isotope effect is present.
(M = Li, Na, and K) has been carried out to study how the structure and dynamics of low-barrier hydrogen-bonded Zundel anion, HO, can be affected by the counter alkali metal cation, M. Our simulation predicts that the quantum proton transfer in Zundel anion can be strongly coupled to the motion of counter cation located nearby. A smaller cation can induce larger structural distortion of the Zundel anion fragment making the proton transfer barrier higher, and hence lowers the vibration excitation energy. It is also argued that a large H/D isotope effect is present.
 path integral hybrid Monte Carlo based on the fourth-order Trotter expansion; Application to fluoride ion-water cluster
path integral hybrid Monte Carlo based on the fourth-order Trotter expansion; Application to fluoride ion-water clusterSuzuki, Kimichi*; Tachikawa, Masanori*; Shiga, Motoyuki
Journal of Chemical Physics, 132(14), p.144108_1 - 144108_7, 2010/04
Times Cited Count:34 Percentile:75.13(Chemistry, Physical)We propose an efficient path integral hybrid Monte Carlo (PIHMC) method based on fourth-order Trotter expansion. Here, the second-order effective force is employed to generate short trial trajectories to avoid computationally expensive Hessian matrix, while the final acceptance is judged based on fourth-order effective potential. The computational performance of our PIHMC scheme is compared with that of conventional PIHMC and PIMD methods based on second- and fourth-order Trotter expansions. Our method is applied to 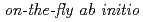 PIHMC calculation of fluoride ion-water complexes, F
PIHMC calculation of fluoride ion-water complexes, F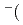 HO) and F(DO), at ambient temperature, particularly focusing on the geometrical isotope effect.
HO) and F(DO), at ambient temperature, particularly focusing on the geometrical isotope effect.
path integral simulationShiga, Motoyuki; Suzuki, Kimichi*; Tachikawa, Masanori*
Journal of Chemical Physics, 132(11), p.114104_1 - 114104_7, 2010/03
Times Cited Count:17 Percentile:50.3(Chemistry, Physical)The 1H NMR chemical shift of deprotonated water dimer, H O, has been studied by ab initio path integral simulation. The simulation predicts that the isotropic shielding of hydrogen-bonded proton increases as a function of temperature. This change is about an order of magnitude larger than that of the non-hydrogen-bonded proton. It is concluded that this is caused by the significant difference in the quantum distribution of proton at high and low temperatures in the low barrier hydrogen bond.
O, has been studied by ab initio path integral simulation. The simulation predicts that the isotropic shielding of hydrogen-bonded proton increases as a function of temperature. This change is about an order of magnitude larger than that of the non-hydrogen-bonded proton. It is concluded that this is caused by the significant difference in the quantum distribution of proton at high and low temperatures in the low barrier hydrogen bond.
Suzuki, Kimichi*; Shiga, Motoyuki; Tachikawa, Masanori*
Journal of Chemical Physics, 129(144), p.144310_1 - 144310_8, 2008/10
Times Cited Count:48 Percentile:84.57(Chemistry, Physical)Path integral molecular dynamics simulation based on 4th order Trotter expansion has been performed to elucidate the geometrical isotope effect of water dimer anion, HO, DO, and TO, at different temperatures from 50 K to 600 K. At low temperatures below 200 K the hydrogen-bonded hydrogen nucleus is near the center of two oxygen atoms with mostly O...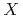 ...O geometry (where = H, D, or T), while at high temperatures above 400 K hydrogen becomes more delocalized showing the coexistence between O...-O and O-...O. The OO distance tends to be shorter as the isotopomer is heavier at low temperatures, while this ordering becomes opposite at high temperatures. It is concluded that the coupling between the OO stretching mode and proton transfer modes is a key to understand such a temperature dependence of hydrogen-bonded structure.
...O geometry (where = H, D, or T), while at high temperatures above 400 K hydrogen becomes more delocalized showing the coexistence between O...-O and O-...O. The OO distance tends to be shorter as the isotopomer is heavier at low temperatures, while this ordering becomes opposite at high temperatures. It is concluded that the coupling between the OO stretching mode and proton transfer modes is a key to understand such a temperature dependence of hydrogen-bonded structure.
