


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Li, H.*; Sakuraba, Shun; Chandrasekaran, A.*; Yang, L.-W.*
Journal of Chemical Information and Modeling, 54(8), p.2275 - 2285, 2014/08
Times Cited Count:16 Percentile:69.28(Chemistry, Medicinal)We provide evidence supporting that protein-protein and protein-ligand docking poses are functions of protein shape and intrinsic dynamics. Over sets of 68 protein-protein complexes and 240 non-homologous enzymes, we recognize common predispositions for binding sites to have minimal vibrations and angular momenta while two interacting proteins orient so as to maximize the angle between their rotation/bending axes (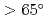 ). The findings are then used to define quantitative criteria to filter out docking decoys less likely to be the near-native poses, hence the chances to find near-native hits can be doubled. With the novel approach to partition a protein into `domains' of robust but disparate intrinsic dynamics, 90% of catalytic residues in enzymes can be found within the first 50% of the residues closest to the interface of these dynamics domains. The results suggest an anisotropic rather than isotropic distribution of catalytic residues near the mass centers of enzymes.
). The findings are then used to define quantitative criteria to filter out docking decoys less likely to be the near-native poses, hence the chances to find near-native hits can be doubled. With the novel approach to partition a protein into `domains' of robust but disparate intrinsic dynamics, 90% of catalytic residues in enzymes can be found within the first 50% of the residues closest to the interface of these dynamics domains. The results suggest an anisotropic rather than isotropic distribution of catalytic residues near the mass centers of enzymes.
