


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Lobzenko, I.; Tsuru, Tomohito; Shiihara, Yoshinori*; Iwashita, Takuya*
Materials Research Express (Internet), 10(8), p.085201_1 - 085201_12, 2023/08
Times Cited Count:0 Percentile:0(Materials Science, Multidisciplinary)Unlike alloys with a crystal lattice, metallic glasses (MG) do not possess distinctive defects but demonstrate a highly heterogeneous response to mechanical deformation, even in near-elastic regimes. The difficulties in describing such non-uniform behavior hamper the prediction of the mechanical properties of MGs. We apply first-principles calculations of atomic stress in CuZr MG to reveal its response to shear strain. That approach allows one to probe such parameters as displacement vector, charge transfer, or change in chemical bonds on the lowest atomic level. We find correlations between the mentioned parameters and show the importance of atomic von Mises stress in the comprehensive description of the mechanical state of a glassy system.
Mori, Hideki*; Tsuru, Tomohito; Okumura, Masahiko; Matsunaka, Daisuke*; Shiihara, Yoshinori*; Itakura, Mitsuhiro
Physical Review Materials (Internet), 7(6), p.063605_1 - 063605_8, 2023/06
Times Cited Count:0 Percentile:0(Materials Science, Multidisciplinary)The introduction of obstacles (e.g., precipitates) for controlling dislocation motion in molecular structures is a prevalent method for designing the mechanical strength of metals. Owing to the nanoscale size of the dislocation core (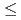 1 nm), atomic modeling is required to investigate the interactions between the dislocation and obstacles. However, conventional empirical potentials are not adequately accurate, in contrast to the calculations based on density functional theory (DFT). Therefore, the atomic-level details of the interactions between the dislocations and obstacles remain unclarified. To this end, this study applied an artificial neural network (ANN) framework to construct an atomic potential by leveraging the high accuracy of DFT. Using the constructed ANN potential, we investigated the dynamic interaction between the
1 nm), atomic modeling is required to investigate the interactions between the dislocation and obstacles. However, conventional empirical potentials are not adequately accurate, in contrast to the calculations based on density functional theory (DFT). Therefore, the atomic-level details of the interactions between the dislocations and obstacles remain unclarified. To this end, this study applied an artificial neural network (ANN) framework to construct an atomic potential by leveraging the high accuracy of DFT. Using the constructed ANN potential, we investigated the dynamic interaction between the 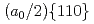 edge dislocation and obstacles in BCC iron. When the dislocation crossed the void, an ultrasmooth and symmetric half-loop was observed for the bowing-out dislocation. Except for the screw dislocation, the Peierls stress of all the dislocations predicted using the ANN was less than 100 MPa. More importantly, the results confirmed the formation of an Orowan loop in the interaction between a rigid sphere and dislocation. Furthermore, we discovered a phenomenon in which the Orowan loop disintegrated into two small loops during its interaction with the rigid sphere and dislocation.
edge dislocation and obstacles in BCC iron. When the dislocation crossed the void, an ultrasmooth and symmetric half-loop was observed for the bowing-out dislocation. Except for the screw dislocation, the Peierls stress of all the dislocations predicted using the ANN was less than 100 MPa. More importantly, the results confirmed the formation of an Orowan loop in the interaction between a rigid sphere and dislocation. Furthermore, we discovered a phenomenon in which the Orowan loop disintegrated into two small loops during its interaction with the rigid sphere and dislocation.
Lobzenko, I.; Wei, D.*; Itakura, Mitsuhiro; Shiihara, Yoshinori*; Tsuru, Tomohito
Results in Materials (Internet), 17, p.100364_1 - 100364_7, 2023/03
High-entropy alloys (HEAs) have received attention for their excellent mechanical and thermodynamic properties. A recent study revealed that Co-free face-centered cubic HEAs carried a potential to improve strength and ductility, which is of high importance for nuclear materials. Here, we implemented first-principles calculations to explore the fundamental mechanism of improving mechanical properties in Co-free HEA. We found that the local lattice distortion of Co-free HEA is more significant than that of the well-known Cantor alloy. In addition, the short-range order formation in Co-free HEA caused highly fluctuated stacking fault energy. Thus, the significant local lattice distortion and the non-uniform solid solution states composed of low- and high-stacking fault regions contribute to improving strength and ductility.
Shiihara, Yoshinori*; Kanazawa, Ryosuke*; Matsunaka, Daisuke*; Lobzenko, I.; Tsuru, Tomohito; Koyama, Masanori*; Mori, Hideki*
Scripta Materialia, 207, p.114268_1 - 114268_4, 2022/01
Times Cited Count:12 Percentile:73.14(Nanoscience & Nanotechnology)This study reports grain boundary (GB) energy calculations for 46 symmetric-tilt GBs in  -iron using molecular mechanics based on an artificial neural network (ANN) potential and compares the results with calculations based on the density functional theory (DFT), the embedded atom method (EAM), and the modified EAM (MEAM). The results by the ANN potential are in excellent agreement with those of the DFT (5% on average), while the EAM and MEAM significantly differ from the DFT results (about 27% on average). In a uniaxial tensile calculation of GB, the ANN potential reproduced the brittle fracture tendency of the GB observed in the DFT while the EAM and MEAM mistakenly showed ductile behaviors. These results demonstrate the effectiveness of the ANN potential in calculating grain boundaries of iron, which is in high demand in modern industry.
-iron using molecular mechanics based on an artificial neural network (ANN) potential and compares the results with calculations based on the density functional theory (DFT), the embedded atom method (EAM), and the modified EAM (MEAM). The results by the ANN potential are in excellent agreement with those of the DFT (5% on average), while the EAM and MEAM significantly differ from the DFT results (about 27% on average). In a uniaxial tensile calculation of GB, the ANN potential reproduced the brittle fracture tendency of the GB observed in the DFT while the EAM and MEAM mistakenly showed ductile behaviors. These results demonstrate the effectiveness of the ANN potential in calculating grain boundaries of iron, which is in high demand in modern industry.
Adachi, Nozomu*; Matsuo, Yasutaka*; Todaka, Yoshikazu*; Fujimoto, Mikiya*; Hino, Masahiro*; Mitsuhara, Masatoshi*; Oba, Yojiro; Shiihara, Yoshinori*; Umeno, Yoshitaka*; Nishida, Minoru*
Tribology International, 155, p.106781_1 - 106781_9, 2021/03
Times Cited Count:7 Percentile:58.99(Engineering, Mechanical) precipitate in Al-Mg-Zn alloys
precipitate in Al-Mg-Zn alloysTsuru, Tomohito; Yamaguchi, Masatake; Ebihara, Kenichi; Itakura, Mitsuhiro; Shiihara, Yoshinori*; Matsuda, Kenji*; Toda, Hiroyuki*
Computational Materials Science, 148, p.301 - 306, 2018/06
Times Cited Count:44 Percentile:82.01(Materials Science, Multidisciplinary)Hydrogen embrittlement susceptibility of high strength 7xxx series Al alloys has been recognized as the critical issues in the practical use of Al alloys. Focusing on the interface between MgZn precipitates and an Al matrix, which is considered as one of the important segregation sites in these alloys, we investigated the stable 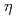 -MgZn-Al interface, and the possible hydrogen trap sites in MgZn and at the -MgZn-Al interface via first-principles calculation. Most of the interstitial sites inside the MgZn crystal were not possible trap sites because their energy is relatively higher than that of other trap sites. The trap energy of the most favorable site at the -MgZn-Al is approximately -0.3 eV/H, which is more stable that of the interstitial site at the grain boundary. The interface between MgZn and Al is likely to be a possible trap site in Al alloys.
-MgZn-Al interface, and the possible hydrogen trap sites in MgZn and at the -MgZn-Al interface via first-principles calculation. Most of the interstitial sites inside the MgZn crystal were not possible trap sites because their energy is relatively higher than that of other trap sites. The trap energy of the most favorable site at the -MgZn-Al is approximately -0.3 eV/H, which is more stable that of the interstitial site at the grain boundary. The interface between MgZn and Al is likely to be a possible trap site in Al alloys.
Tsuru, Tomohito; Lobzenko, I.; Shiihara, Yoshinori*; Wei, D.*; Yamashita, Shinichiro; Itakura, Mitsuhiro; 10 of others*
no journal, ,
High entropy alloys (HEAs) are chemically complex single- or multi-phase alloys with crystal structures. There are no major components but five or more elements are included with near equiatomic fraction. In such a situation, deformation behavior can no longer be described by conventional solid solution strengthening model. Some HEAs, indeed, show higher strengthening behavior and anomalous slip. However, the mechanisms of these features have yet to be understood. Dislocation structure and motion should be the key to identify the unique feature of mechanical properties of HEAs. In the present study, we investigated the core structure of dislocations in body centered cubic (BCC) HEAs using density functional theory (DFT) calculations. The Random structure and ZrNbTaTiHf and the SRO structure obtained from the 800 K MC calculation in two BCC-HEA MoNbTaVW was prepared. Then, the energy distribution when the dislocation dipoles were introduced at 135 sites were calculated. We found that the dislocation formation energy is smaller in ZrNbTaTiHf, which has a large difference in MSAD and a large lattice distortion.
Mori, Hideki*; Itakura, Mitsuhiro; Okumura, Masahiko; Shiihara, Yoshinori*; Matsunaka, Daisuke*
no journal, ,
no abstracts in English
Lobzenko, I.; Shiihara, Yoshinori*; Tsuru, Tomohito
no journal, ,
High-entropy alloys (HEA) are excellent structural materials due to their promising mechanical properties. Works on body-centered cubic (BCC) HEAs show increased ductility if group 4 elements are present in the composition. Theoretical studies of that effect by first-principles modeling are complicated by the essential randomness of HEA atomic structure, which requires large systems. To achieve high accuracy in classical molecular dynamics we have developed interatomic potentials using machine learning of artificial neural networks (we refer to them as ANN potentials). We present in the current work results for two medium-entropy alloys (MEA): MoNbTa and ZrNbTa. Comparison of basic mechanical properties show decrease of bulk modulus and elastic constants if Mo is substituted with group 4 element Zr. Edge and screw dislocations are studied. Classical modelling allows construction of big calculation cells, that prevents self-interaction of the dislocation core due to long-range stress field. Moreover, big cells ensures better randomness of alloys, which is vital in simulations of HEA and MEA mechanical properties. Screw dislocation movement is induced by applying shear strain. In case of edge dislocation the shape and energy is studied in the process of migration of the dislocation core between two adjacent easy core configurations. In this way the Peierls barrier is calculated. Results for two MEA are compared to elucidate the role of group 4 element. Finally, to understand the stress field of dislocations we employ atomic stress calculation scheme in the framework of ANN potentials. Atomic stress calculations is possible based on virial stress definition due to the fact that atomic energy in the ANN scheme ultimately depends on pair distances between atoms.
Lobzenko, I.; Tsuru, Tomohito; Shiihara, Yoshinori*; Mori, Hideki*
no journal, ,
High-entropy alloys (HEA) exhibit excellent mechanical properties, which makes them good candidates for structural materials. Works on body-centered cubic (BCC) HEAs show increased ductility if HCP elements are present in the composition. Origins of that effect could be studied in computer experiments, but first-principles modeling is complicated by the essential randomness of HEA atomic structure, which requires large systems. To achieve high accuracy in classical molecular dynamics we apply the technique of machine learning for interatomic potentials development. Current work focuses on the mechanical properties of two medium-entropy alloys (MEA): MoNbTa and ZrNbTa. Interatomic potentials were built for these two alloys using artificial neural networks (we refer to them as ANN potentials). Our study reveals a drastic change in the basic properties of the material when Mo is substituted with HCP element Zr. In particular, the alloy with Zr has decreased bulk modulus and elastic constants. The change in C11 and C12 elastic constants shows that the material comes closer to the elastic instability region. We have also studied the shape of the screw dislocation core in the two MEA. Results show that the non-compact core shape in the ZrNbTa alloy has a larger width.
Lobzenko, I.; Shiihara, Yoshinori*; Mori, Hideki*; Matsunaka, Daisuke*; Tsuru, Tomohito
no journal, ,
Recently methods of machine learning have become an important part of materials science. Particularly, interatomic potentials built using such methods demonstrate accuracy of geometrical characteristics of materials approaching the accuracy of first-principle calculations. In our work we use artificial neural networks to build potentials, and therefore they are called ANN potentials. We focus on the atomic stress, one of the important properties of materials, which can be calculated in classical approximation. Analysis of microscopic stress can be applied to any type of nonuniform system (such as defects in bulk, two-dimensional crystals, molecules assemblies, etc.). We show how the central-force decomposition (CFD) scheme can be used in the framework of ANN potentials for the derivation of atomic stress tensor. It is important to use CFD due to the fact that the symmetry of the stress tensor may be broken in other schemes. Finally, we calculate atomic stress distributions near surfaces of pure Al with different orientations. It is known from first-principle studies that there is a charge oscillations near Al surfaces, however it cannot be captured by existing interatomic potentials. Our results, obtained with the new ANN potential, show oscillations of atomic stress near Al surface. Even though our potential was fitted to only energies of Al structures (calculated in quantum-mechanics approximation), we attribute the atomic stress oscillations to the charge distribution of the real system. Charge oscillations are affecting total energies of structures, and therefore are implicitly included in the data set, which we use for building the potential.
Lobzenko, I.; Tsuru, Tomohito; Shiihara, Yoshinori*; Iwashita, Takuya*
no journal, ,
Revealing the origin of the mechanical properties of metallic glasses (MG) is a long-standing problem. MGs respond to the external strain with the activation of collective atomic motion, but the triggers of such motions are not revealed yet, in contrast to the well-defined dislocations in crystals. To study this collective atomic response in detail, we use the atomic stress calculations in the first-principles framework. Four small random Cu50%Zr50% structures were prepared and put under the strain from 0.5 to 8.0%. The stress response is shown separately for Cu and Zr. We analyze the system's transformation between the affine and relaxed states and find a significant deviation from elastic behavior. As the atomic von Mises stress change indicates, the xy shear strain invokes atomic stress response in other shear components. Other local parameters, such as charge transfer, atomic displacements, and atomic strain, are also discussed.
Lobzenko, I.; Shiihara, Yoshinori*; Mori, Hideki*; Matsunaka, Daisuke*; Tsuru, Tomohito
no journal, ,
Refractory multi-component alloys (MCA) form an important class of materials with high potential for use in severe conditions. One of the main problems hindering the application of these alloys is the low ductility inherited from the body-centred cubic (BCC) crystal structure. Dislocation motion is the factor significantly influencing the ductility of the material, so a comprehensive understanding of the dislocation dynamics in refractory MCAs should be achieved to pave the way for designing refractory alloys with increased ductility. To achieve high accuracy in classical molecular dynamics simulations of dislocation motion, we apply the technique of machine learning (ML) for interatomic potential development. It is known that alloys with hexagonal closed-packed (HCP) elements such as Zr exhibit higher ductility, which is why two medium-entropy alloys, MoNbTa and ZrNbTa, were chosen to study the influence of elements' constitution on dislocations dynamics. The inter-atomic potentials for MCAs built using ML need a specific dataset. In the process of the potential development, we identify which structures contribute to a better quality of materials' mechanical properties prediction by the potentials. Results of the simulations have shown qualitative and quantitative differences between the two alloys under study. One example of that difference can be seen in the shapes of the screw dislocation core. In contrast to MoNbTa, ZrNbTa demonstrates a non-compact core with an extension on a (110) plane.
