


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Suzuki, Kento*; Miyazaki, Takaaki*; Takayanagi, Toshiyuki*; Shiga, Motoyuki
Physical Chemistry Chemical Physics, 20(41), p.26489 - 26499, 2018/11
Times Cited Count:1 Percentile:3.34(Chemistry, Physical)The direct photoionization of pure helium clusters and its subsequent short-time process have been studied by path integral molecular dynamics (PIMD) and ring-polymer molecular dynamics (RPMD) simulations. The PIMD simulations reproduced the experimental ionization spectra with a broad and asymmetric nature, which can be ascribed to the inhomogeneity of the energy levels of He atoms. From the RPMD simulations, it is found that the ionized helium cluster in the highly excited state brings about fast electronic state relaxation via nonadiabatic charge transfer and subsequently slow structural relaxation.
Ozama, Eiki*; Adachi, Sadia*; Takayanagi, Toshiyuki*; Shiga, Motoyuki
Chemistry; A European Journal, 24(48), p.12716 - 12721, 2018/08
Times Cited Count:5 Percentile:20.11(Chemistry, Multidisciplinary)The structures of trivalent actinium cation in helium clusters (Ac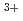 He
He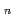 ) have been studied by quantum path integral molecular dynamics simulations with different cluster sizes,
) have been studied by quantum path integral molecular dynamics simulations with different cluster sizes, 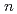 = 18-200. The nuclear quantum effect of helium atoms plays an important role in the vibrational amplitude of the Ac-He complex at low temperatures (1-3 K) where the complex is stable. We found that the coordination number of helium atoms comprising the first solvation shell can be as high as eighteen. In this case, the helium atoms are arranged in D
= 18-200. The nuclear quantum effect of helium atoms plays an important role in the vibrational amplitude of the Ac-He complex at low temperatures (1-3 K) where the complex is stable. We found that the coordination number of helium atoms comprising the first solvation shell can be as high as eighteen. In this case, the helium atoms are arranged in D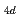 symmetry. The Ac-He
symmetry. The Ac-He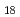 complex becomes more rigid as the cluster increases in sizes, implying that it becomes more stable. The simulation results are based on an accurate description of the Ac-He interaction using relativistic ab initio calculations.
complex becomes more rigid as the cluster increases in sizes, implying that it becomes more stable. The simulation results are based on an accurate description of the Ac-He interaction using relativistic ab initio calculations.
Seki, Yusuke*; Takayanagi, Toshiyuki*; Shiga, Motoyuki
Physical Chemistry Chemical Physics, 19(21), p.13798 - 13806, 2017/06
Times Cited Count:8 Percentile:35.36(Chemistry, Physical)Ring-polymer molecular dynamics (RPMD) simulations have been performed to understand the photoexcitation dynamics of the Ag atom embedded in a low-temperature cluster consisting of 500 helium atoms, after the electronic excitation of the Ag atom. Along the RPMD trajectory the time evolution of the electronic wavefunction within the spin-orbit  P manifold is calculated, whereby the time-dependent Schr
P manifold is calculated, whereby the time-dependent Schr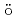 dinger equation and the RPMD equation of motion are coupled,
dinger equation and the RPMD equation of motion are coupled, 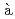 la Ehrenfest mean field approach. It is found from the simulations that the Ag atom is mostly ejected from the helium cluster with the average time of 100 ps after photoexcitation. The average velocity of the ejected Ag atom is estimated to be 60-70 m/s. These results are qualitatively in line with previous experimental findings.
la Ehrenfest mean field approach. It is found from the simulations that the Ag atom is mostly ejected from the helium cluster with the average time of 100 ps after photoexcitation. The average velocity of the ejected Ag atom is estimated to be 60-70 m/s. These results are qualitatively in line with previous experimental findings.
Minoshima, Yusuke*; Seki, Yusuke*; Takayanagi, Toshiyuki*; Shiga, Motoyuki
Chemical Physics, 472, p.1 - 8, 2016/06
Times Cited Count:2 Percentile:6.19(Chemistry, Physical)The dynamical process of electron attachment to a guanine-cytosine pair in the normal (h-GC) and deuterated (d-GC) forms has been studied theoretically by semiclassical ring-polymer molecular dynamics (RPMD) simulations using the empirical valence bond model. The initially formed dipole-bound anion is converted rapidly to the valence-bound anion within about 0.1 ps in both h-GC and d-GC. However, the subsequent proton transfer in h-GC occurs with a rate five times greater than the deuteron transfer in d-GC. The change of rates with isotopic substitution and temperature variation in the RPMD simulations are quantitatively and qualitatively different from those in the classical molecular dynamics (MD) simulations, demonstrating the importance of nuclear quantum effects on the dynamics of this system.
Honda, Tomohiro*; Minoshima, Yusuke*; Yokoi, Yuki*; Takayanagi, Toshiyuki*; Shiga, Motoyuki
Chemical Physics Letters, 625, p.174 - 178, 2015/04
Times Cited Count:5 Percentile:19.29(Chemistry, Physical)Electron attachment dynamics to the guanine-cytosine (G-C) base pair in the gas phase isstudied using DFT and molecular dynamics. The potential energy surface of the G-C anion isconstructed with the empirical-valence-bond method using force-field information obtained from long-range corrected DFT calculations. Ring-polymer molecular dynamics simulations predict that theinitial dipole-bound anion readily converts into the valence-bound anion within 0.1 ps and proton-transfer occurs subsequently within 10 ps. The same process was found in classical simulations, buton a much slower time scale. This result suggests that nuclear quantum effects are important inunderstanding DNA damage by low-energy electrons.
 -iron
-ironYoshikawa, Takehiro*; Takayanagi, Toshiyuki*; Kimizuka, Hajime*; Shiga, Motoyuki
Journal of Physical Chemistry C, 116(43), p.23113 - 23119, 2012/11
Times Cited Count:16 Percentile:45.57(Chemistry, Physical)The diffusion coefficients of hydrogen (H) and tritium (T) in -Fe have been computed using two approximate quantum dynamical techniques, i.e. centroid molecular dynamics (CMD) and ring polymer molecular dynamics (RPMD), in the temperature range of T = 100-1000 K using the embedded-atom-method (EAM) potential. It has been found that the RPMD and CMD methods give very similar results. From a further analysis based on quantum transition state theory (centroid density QTST) combined with path integral molecular dynamics (PIMD), it has been clear that there is a crossover between thermal and quantum mechanisms at about T = 500 K and 300 K for H and T diffusions, respectively. The importance of nuclear quantum effects at low temperatures has been illustrated in terms of the effective free energy surface map.
Yoshikawa, Takehiro*; Sugawara, Shuichi*; Takayanagi, Toshiyuki*; Shiga, Motoyuki; Tachikawa, Masanori*
Chemical Physics, 394(1), p.46 - 51, 2012/02
Times Cited Count:18 Percentile:55.59(Chemistry, Physical)Path-integral molecular dynamics simulations have been performed for porphycene and its isotopic variants in order to understand the effect of isotopic substitution of inner protons on the double proton transfer mechanism. Our quantum simulations show that double proton transfer of the unsubstituted porphycene at the room temperature mainly occurs via a so-called concerted mechanism through the second-order saddle point. In addition, we found that both isotopic substitution and temperature significantly affect the double proton transfer mechanism. For example, the contribution of the stepwise mechanism increases with a temperature increase. It has been found that out-of-plane vibrational motions significantly decrease the contribution of the concerted proton transfer mechanism.
Sugawara, Shuichi*; Yoshikawa, Takehiro*; Takayanagi, Toshiyuki*; Shiga, Motoyuki; Tachikawa, Masanori*
Journal of Physical Chemistry A, 115(42), p.11486 - 11494, 2011/09
Times Cited Count:19 Percentile:56.05(Chemistry, Physical)We have carried out path integral molecular dynamics simulations for hydrated sulfuric acid clusters to understand acid-dissociation and hydrogen-bonded structural rearrangement processes in these clusters from a quantum mechanical viewpoint. We have found that the acid dissociation processes, first and second deprotonation, effectively occur in a hydrated cluster with a specific cluster size. The mechanisms of the proton-transfer processes were analyzed in detail and it was found that the distance between O in sulfuric acid and O in the proton-accepting water is playing an important role. We also found that the water coordination number of the proton-accepting water is important in the proton-transfer processes.
Yoshikawa, Takehiro*; Sugawara, Shuichi*; Takayanagi, Toshiyuki*; Shiga, Motoyuki; Tachikawa, Masanori*
Chemical Physics Letters, 496(1-3), p.14 - 19, 2010/08
Times Cited Count:27 Percentile:67.43(Chemistry, Physical)Full-dimensional path-integral molecular dynamics simulations were performed to determine whether the double proton transfer tautomerization of porphycene is a concerted or a stepwise process. It was found that double proton transfer occurs dominantly through the concerted pathway via the second-order saddle point structure and that contribution of the stepwise mechanism increases with a temperature increase. Nuclear quantum effects play essential roles in determining the proton transfer mechanism.
 O)
O)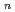 ( = 1-7) clusters on semi-empirical PM6 potential energy surfaces
( = 1-7) clusters on semi-empirical PM6 potential energy surfacesYoshikawa, Takehiro*; Motegi, Haruki*; Kakizaki, Akira*; Takayanagi, Toshiyuki*; Shiga, Motoyuki
Chemical Physics, 365(1-2), p.60 - 68, 2009/12
Times Cited Count:15 Percentile:46.32(Chemistry, Physical)Path-integral molecular dynamics simulations for the hydrogen-bonded glycine-(HO) ( = 1-7) clusters have been carried out using an on-the-fly direct dynamics technique at the semiempiricalPM6 level of theory. In the case of smaller clusters with = 1-3, the simulations show that the clusterstructure takes exclusively the hydrogen-bonded complex between a canonical neutral glycine and a water cluster moiety. In contrast, it was found that proton-exchange processes effectively occur between the COOH carboxylic group and water cluster moiety for = 4-6 clusters although the overall structures are the complex between a neutral glycine and water clusters. In the case of the = 7 cluster, glycine preferentially takes a zwitterionic form having NH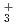 and COO
and COO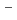 functional groups.
functional groups.
O)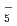 and (DO)
and (DO)Takayanagi, Toshiyuki*; Yoshikawa, Takehiro*; Motegi, Haruki*; Shiga, Motoyuki
Chemical Physics Letters, 482(4-6), p.195 - 200, 2009/12
Times Cited Count:9 Percentile:29.21(Chemistry, Physical)Path-integral molecular dynamics simulations have been performed for water anion clusters,(HO) and (DO), on the basis of a semiempirical one-electron pseudopotential polarization model. Due to larger zero-point vibrational amplitudes for H atoms than that of D atoms, hydrogen-bond lengths in the (HO) cluster are slightly larger than those in (DO). The distribution of the vertical detachment energies for (HO) also show a broader feature than that for (DO). The present simulations thus demonstrate the importance of nuclear quantum effects in water anion clusters.
SO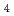
 (HO) (=1-6) on semiempirical PM6 potential surfaces
(HO) (=1-6) on semiempirical PM6 potential surfacesKakizaki, Akira*; Motegi, Haruki*; Yoshikawa, Takehiro*; Takayanagi, Toshiyuki*; Shiga, Motoyuki; Tachikawa, Masanori*
Journal of Molecular Structure; THEOCHEM, 901(1-3), p.1 - 8, 2009/05
Quantum path-integral molecular dynamics simulations for a series of sulfuric acid clusters, HSO(HO)(=1-6), were performed using semiempirical PM6 method. It was found that the acid dissociation probability increases with an increase in the cluster size, and that so-called contact-ion-pair structures are dominant in the proton-dissociated clusters. The importance of nuclear quantum effects in the cluster structures and proton-transfer processes is demonstrated.
O) on a semiempirical potential energy surfaceTakayanagi, Toshiyuki*; Takahashi, Kenta*; Kakizaki, Akira*; Shiga, Motoyuki; Tachikawa, Masanori*
Chemical Physics, 358(3), p.196 - 202, 2009/04
Times Cited Count:16 Percentile:48.94(Chemistry, Physical)Path-integral molecular dynamics simulations for the HCl(HO) cluster have been performed on the ground-state potential energy surface directly obtained on-the-fly from semiempirical PM3-MAIS molecular orbital calculations. It is found that the HCl(HO) cluster has structural rearrangement above the temperature of 300 K showing a liquid-like behavior. Quantum mechanical fluctuation of hydrogen nuclei plays a significant role in structural arrangement processes in this cluster.
Motegi, Haruki*; Kakizaki, Akira*; Takayanagi, Toshiyuki*; Taketsugu, Yuriko*; Taketsugu, Tetsuya*; Shiga, Motoyuki
Chemical Physics, 354(1-3), p.38 - 43, 2008/12
Times Cited Count:12 Percentile:39.25(Chemistry, Physical)Path-integral molecular dynamics simulations have been performed to understand the quantum helium solvation structures in the He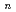 BeO cluster. Our simulations show that one helium atom is strongly bound to BeO to form HeBeO and that the first solvation shell around the HeBeO complex includes roughly 12-14 helium atoms. The present simulations implies that the HeBeO complex could be experimentally detected in large helium clusters using the HENDI technique.
BeO cluster. Our simulations show that one helium atom is strongly bound to BeO to form HeBeO and that the first solvation shell around the HeBeO complex includes roughly 12-14 helium atoms. The present simulations implies that the HeBeO complex could be experimentally detected in large helium clusters using the HENDI technique.
 O) (=2-7) clusters on semiempirical PM6 potential energy surfaces
O) (=2-7) clusters on semiempirical PM6 potential energy surfacesTakayanagi, Toshiyuki*; Yoshikawa, Takehiro*; Kakizaki, Akira*; Shiga, Motoyuki; Tachikawa, Masanori*
Journal of Molecular Structure; THEOCHEM, 869(1-3), p.29 - 36, 2008/11
Molecular dynamics simulations based on semiempirical PM6 method was carried out to study statistical structures of glycine-water clusters. We found that zwitterionic form is more stable than neutral form as the cluster size increases. The difference in water solvation structures around the neutral or zwitterionic glycine form is also discussed.
systemKurosaki, Yuzuru; Takayanagi, Toshiyuki*
Chemical Physics Letters, 406(1-3), p.121 - 125, 2005/04
no abstracts in English
 in irradiated solid
in irradiated solid 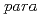 -hydrogen and its decay dynamics; Reinvestigation of quartet electron paramagnetic resonance lines assigned to H
-hydrogen and its decay dynamics; Reinvestigation of quartet electron paramagnetic resonance lines assigned to HKumada, Takayuki; Tachikawa, Hiroto*; Takayanagi, Toshiyuki*
Physical Chemistry Chemical Physics, 7(5), p.776 - 784, 2005/02
Times Cited Count:12 Percentile:38.35(Chemistry, Physical)The quartet ESR lines observed in X-ray irradiated solid parahydrogen, which have previously been assigned to H, are reinvestigated. We have reassigned the quartet lines to H rather than H. Based on the new assignment, previous data have been reinterpreted as follows. The H ion is composed of the collinearly aligned H core at the center and two H rotors at both ends. The ortho-para conversion of H core of H is completed within the time-scale of hours. H diffuses quantum mechanically by the repetition of H + H  H + H reaction, and the diffusion terminates by the reaction with a HD impurity as H + HD H
H + H reaction, and the diffusion terminates by the reaction with a HD impurity as H + HD H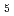 D + H. Finally, we will propose a possible reason why H is produced instead of H
D + H. Finally, we will propose a possible reason why H is produced instead of H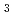 in the irradiated solid H.
in the irradiated solid H.
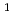 D)+NONO+NO/N+O reaction
D)+NONO+NO/N+O reactionTakayanagi, Toshiyuki
Chemical Physics, 308(3), p.211 - 216, 2005/01
Times Cited Count:6 Percentile:20.38(Chemistry, Physical)no abstracts in English
Takayanagi, Toshiyuki
Chemical Physics, 302(1-3), p.85 - 93, 2004/07
Times Cited Count:12 Percentile:36.82(Chemistry, Physical)no abstracts in English
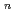 exciplexes
exciplexesTakayanagi, Toshiyuki; Shiga, Motoyuki
Physical Chemistry Chemical Physics, 6(13), p.3241 - 3247, 2004/07
Times Cited Count:29 Percentile:67.82(Chemistry, Physical)no abstracts in English
