


最低励起三重項状態(T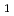 )のポテンシャルエネルギー面(PES)上でのアセトアルデヒドの光分解における水素原子生成チャンネル: CD
)のポテンシャルエネルギー面(PES)上でのアセトアルデヒドの光分解における水素原子生成チャンネル: CD CHO
CHO CD
CD CHO+D(1); CDCHOCDCO+H(2)について、密度汎関数理論(DFT)に基づく直接分子動力学計算を実施した。DFT法はB3LYPで、基底関数はcc-pVDZを用いた。計算の結果、生成した水素原子の平均の運動エネルギーは反応1, 2でそれぞれ18.3, 16.6kcal mol
CHO+D(1); CDCHOCDCO+H(2)について、密度汎関数理論(DFT)に基づく直接分子動力学計算を実施した。DFT法はB3LYPで、基底関数はcc-pVDZを用いた。計算の結果、生成した水素原子の平均の運動エネルギーは反応1, 2でそれぞれ18.3, 16.6kcal mol であった。これらは、実験値[Kang et al., Chem. Phys. Lett. 434 (2007) 6]の半分から2/3の値であった。計算値が実験値に比べてかなり小さい理由は、反応1, 2の生成物側から見た障壁がB3LYP/cc-pVDZレベルでは小さく見積もられていることにある。しかしながら、ここでの計算結果は実験値と定性的には一致しており、これらの反応がTPES上で起こることを支持するものである。
であった。これらは、実験値[Kang et al., Chem. Phys. Lett. 434 (2007) 6]の半分から2/3の値であった。計算値が実験値に比べてかなり小さい理由は、反応1, 2の生成物側から見た障壁がB3LYP/cc-pVDZレベルでは小さく見積もられていることにある。しかしながら、ここでの計算結果は実験値と定性的には一致しており、これらの反応がTPES上で起こることを支持するものである。
Direct density-functional (DFT) molecular-dynamics (MD) calculations have been carried out for the following two hydrogen atom production channels in acetaldehyde photodissociation on the lowest triplet-state (T) potential energy surface (PES): CDCHOCDCHO+D(1); CDCHOCDCO+H(2). The employed DFT method was B3LYP with the cc-pVDZ basis set. The average product hydrogen kinetic energies estimated from the results of the direct DFT MD calculations were 18.3 and 16.6 kcal mol for reactions 1 and 2, respectively, and these were half - two thirds of the previously measured values [Kang et al. Chem. Phys. Lett. 434 (2007) 6]. This is because of the low reverse barrier heights predicted at the B3LYP/cc-pVDZ level. The present results for the product hydrogen kinetic energies, however, agree qualitatively with the experimental measurements and strongly supports the mechanisms taking place on the T PES.
発表言語 |
: | English |
|---|---|---|
掲載資料名 |
: | |
巻 |
: | 850 |
号 |
: | 1-3 |
ページ数 |
: | p.9 - 16 |
発行年月 |
: | 2008/02 |
キーワード |
: | no keyword |
論文URL |
: |
|
|---|---|---|
論文解説記事 |
: |
Access |
: |
- Accesses |
|---|---|---|
InCites™ |
: |
パーセンタイル:29.05 分野:Chemistry, Physical |
Altmetrics |
: |
[CLARIVATE ANALYTICS], [WEB OF SCIENCE], [HIGHLY CITED PAPER & CUP LOGO] and [HOT PAPER & FIRE LOGO] are trademarks of Clarivate Analytics, and/or its affiliated company or companies, and used herein by permission and/or license.
