


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
黒崎 譲*; 横山 啓一
Universe (Internet), 5(5), p.109_1 - 109_15, 2019/05
被引用回数:0 パーセンタイル:0.00(Astronomy & Astrophysics)二原子ハロゲン化アルカリ分子の振動回転励起に関して、最適制御理論に基づいて最適レーザー電場波形を計算した。今回は2光子過程の効果を計算に取り入れるために分子と電場の相互作用ハミルトニアンに分極率の項を追加した。その結果、弱い電場強度では従来と同じ1光子過程のみが関与する最適波形が得られた。一方、比較的強い電場強度では2光子過程と1光子過程が同時に寄与する電場波形が得られた。これらの結果は2光子過程の効果を最適制御計算にうまく取り込めていることを示唆している。これにより、より現実的な計算が可能になった。
黒崎 譲*; 横山 啓一
Chemical Physics, 493, p.183 - 193, 2017/08
被引用回数:5 パーセンタイル:19.36(Chemistry, Physical)塩化リチウム分子の同位体選択的振動回転励起の最適パルス波形を求めた。振動回転が基底準位にあるLiCl-35とLiCl-37の混合分子集団にレーザーパルスを照射し、LiCl-35(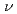 =0,
=0, 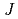 =0)とLiCl-37(=1, =1)を生成する最適波形を最適制御理論により計算した。その結果、回転遷移だけを利用する方が効率よく選択励起を起こすことができることが示された。
=0)とLiCl-37(=1, =1)を生成する最適波形を最適制御理論により計算した。その結果、回転遷移だけを利用する方が効率よく選択励起を起こすことができることが示された。
黒崎 譲; Ho, T.-S.*; Rabitz, H.*
Chemical Physics, 469-470, p.115 - 122, 2016/05
被引用回数:2 パーセンタイル:5.44(Chemistry, Physical)The prospect of performing the open  cyclic ozone isomerization has attracted much research attention. Here we explore this consideration theoretically by performing quantum optimal control calculations to demonstrate the important role that excited-state dissociation channels could play in the isomerization transformation. In the calculations we use a three-state, one-dimensional dynamical model constructed from the lowest five
cyclic ozone isomerization has attracted much research attention. Here we explore this consideration theoretically by performing quantum optimal control calculations to demonstrate the important role that excited-state dissociation channels could play in the isomerization transformation. In the calculations we use a three-state, one-dimensional dynamical model constructed from the lowest five 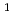 A' potential energy curves obtained with high-level
A' potential energy curves obtained with high-level  calculations. Besides the laser field-dipole couplings between all three states, this model also includes the diabatic coupling between the two excited states at an avoided crossing leading to competing dissociation channels that can further hinder the isomerization process. The present three-state optimal control simulations examine two possible control pathways previously considered in a two-state model, and reveal that only one of the pathways is viable, achieving a robust
calculations. Besides the laser field-dipole couplings between all three states, this model also includes the diabatic coupling between the two excited states at an avoided crossing leading to competing dissociation channels that can further hinder the isomerization process. The present three-state optimal control simulations examine two possible control pathways previously considered in a two-state model, and reveal that only one of the pathways is viable, achieving a robust  95% yield to the cyclic target in the three-state model.
95% yield to the cyclic target in the three-state model.
黒崎 譲
しょうとつ, 12(4), p.114 - 125, 2015/07
近年の量子制御研究において、dynamic Stark効果(DSE)が重要キーワードの一つとなっている。DSEは静電場によるStark効果の振動電場への拡張バージョンといえるが、Stark効果自体はその発見から既に一世紀以上が経過しており目新しいものではない。しかしながら、最近のレーザー技術の飛躍的な進歩により、これまで未知であったDSEの数々の興味深い側面が明らかにされるにつけ、制御研究におけるDSEの重要性はますます高まってきている。本稿では、化学物理学におけるDSEによる量子制御研究の現状を概説するとともに、筆者らの最近の研究成果についても紹介する。
市原 晃; 松岡 雷士*; 黒崎 譲; 横山 啓一
Optical Review, 22(1), p.153 - 156, 2015/02
被引用回数:6 パーセンタイル:32.35(Optics)気体二原子分子を光学パルスにより同位体選択的に振動回転励起させる方法を提案する。この方法では、指定した同位体分子の回転状態をテラヘルツ周波数コムにより励起させる。更に、回転励起した分子を別のパルスでP枝中の基本遷移を通して振動励起させる。提案した同位体選択的励起法の有効性を実証するため、70Kで熱平衡にあるLiCl分子集団に対してコンピューターシミュレーションを実施した。計算には量子力学の基づく緊密結合法を使用し、光学パルスと分子の相互作用はパルス電場と分子の遷移双極子モーメントの積で近似した。その結果、これら二種類のパルスを用いて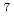 Li
Li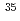 ClとLi
ClとLi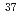 ClのうちLiCl集団を選択的に振動励起できることを確認した。
ClのうちLiCl集団を選択的に振動励起できることを確認した。
黒崎 譲; 赤木 浩; 横山 啓一
Physical Review A, 90(4), p.043407_1 - 043407_9, 2014/10
被引用回数:4 パーセンタイル:23.30(Optics)We theoretically propose a new control scheme of temporal wave-packet separation for oriented molecules, based on nonresonant dynamic Stark effect (DSE) in the dipole limit. In the scheme linearly-polarized single-cycle THz pulses are employed as control fields. In this work the proposed scheme is applied to the temporal wave-packet separation of the binary mixture of alkali halide isotopologues, 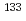 CsI and
CsI and 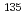 CsI. We assume that before applying the control pulse one of the isotopologues is oriented along the field polarization direction and the other along the opposite direction and then they are electronically excited from the ground-state PEC to the excited-state counterpart. Numerical wave-packet propagations reveal that a THz pulse yields a temporal wave-packet separation of about 2 ps between the two isotopologue photodissociations.
CsI. We assume that before applying the control pulse one of the isotopologues is oriented along the field polarization direction and the other along the opposite direction and then they are electronically excited from the ground-state PEC to the excited-state counterpart. Numerical wave-packet propagations reveal that a THz pulse yields a temporal wave-packet separation of about 2 ps between the two isotopologue photodissociations.
市原 晃; 松岡 雷士; 黒崎 譲; 横山 啓一
JPS Conference Proceedings (Internet), 1, p.013093_1 - 013093_4, 2014/03
塩化リチウム(LiCl)分子の同位体選択的解離過程を、量子力学計算に基づき調べた。基底振動回転状態(v=0, J=0)にあるLiCl及びLiClに対して、スペクトル周波数をLiClの回転遷移周波数に一致させた光周波数コムを照射することにより、LiClだけを選択的に高回転状態に励起できた。コム照射中の分子の振動回転状態分布は、緊密結合法により求めた。コム照射後の第2パルスによるLiClの解離過程は、波束法を用いて計算した。その際、緊密結合計算で得られた振幅を波束計算の入力として使用した。解離確率は減衰関数を用いて波束を吸収させることにより見積もった。その結果、設定した光学パルスによってLiClの約60%が解離した。LiClの解離確率は2%未満であった。
黒崎 譲; Ho, T.-S.*; Rabitz, H.*
Journal of Chemical Physics, 140(8), p.084305_1 - 084305_11, 2014/02
被引用回数:6 パーセンタイル:20.83(Chemistry, Physical)We construct a two-state one-dimensional reaction-path model for ozone open cyclic isomerization dynamics. The model is based on the intrinsic reaction coordinate connecting the cyclic and open isomers with the O + O asymptote on the ground-state
+ O asymptote on the ground-state 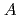 ' potential energy surface obtained with the high-level method. Using this two-state model time-dependent wave packet optimal control simulations are carried out. Two possible pathways are identified along with their respective band-limited optimal control fields; for pathway 1 the wave packet initially associated with the open isomer is first pumped into a shallow well on the excited electronic state potential curve and then driven back to the ground electronic state to form the cyclic isomer, whereas for pathway 2 the corresponding wave packet is excited directly to the primary well of the excited state potential curve. The simulations reveal that the optimal field for pathway 1 produces a final yield of nearly 100% with substantially smaller intensity than that obtained in a previous study using a single-state one-dimensional model. Pathway 2, due to its strong coupling to the dissociation channel, is less effective than pathway 1. The simulations also show that nonlinear field effects due to molecular polarizability and hyperpolarizability are small for pathway 1 but could become significant for pathway 2 because much higher field intensity is involved in the latter.
' potential energy surface obtained with the high-level method. Using this two-state model time-dependent wave packet optimal control simulations are carried out. Two possible pathways are identified along with their respective band-limited optimal control fields; for pathway 1 the wave packet initially associated with the open isomer is first pumped into a shallow well on the excited electronic state potential curve and then driven back to the ground electronic state to form the cyclic isomer, whereas for pathway 2 the corresponding wave packet is excited directly to the primary well of the excited state potential curve. The simulations reveal that the optimal field for pathway 1 produces a final yield of nearly 100% with substantially smaller intensity than that obtained in a previous study using a single-state one-dimensional model. Pathway 2, due to its strong coupling to the dissociation channel, is less effective than pathway 1. The simulations also show that nonlinear field effects due to molecular polarizability and hyperpolarizability are small for pathway 1 but could become significant for pathway 2 because much higher field intensity is involved in the latter.
市原 晃; 松岡 雷士*; 黒崎 譲; 横山 啓一
Chinese Journal of Physics, 51(6), p.1230 - 1240, 2013/12
光周波数コムによる二原子分子の回転状態遷移の確率を定式化した。分子を剛体回転子で近似し、コムのスペクトル周波数を分子の回転遷移周波数と一致させた場合の確率式を誘導した。相互作用は、コムによる電場と分子の遷移双極子モーメントの積で定義した。定式化はデルタキックトローター模型に基づく。異なる時間におけるデルタ関数の直行性を仮定し、スペクトル分解法を用いて解析式を導いた。得られた確率振幅は、第一種ベッセル関数に対応する項と、それ以外の振動関数の部分に分離できることを示した。この確率振幅から得られる遷移確率の時間依存性は、階段関数として表現される。解析式の有効性を証明するため、コム場中のCsI分子に対する回転状態の時間変化を数値計算し、解析式から見積もられる値と比較した。その結果、この解析式により数値計算結果が再現できることを確認した。
市原 晃; 松岡 雷士; 黒崎 譲; 横山 啓一
Proceedings of 10th Conference on Lasers and Electro-Optics Pacific Rim and 18th OptoElectronics and Communications Conference and Photonics in Switching 2013 (CLEO-PR & OECC/PS 2013) (USB Flash Drive), 2 Pages, 2013/06
光周波数コムにより、塩化リチウム分子を同位体選択的に回転励起させるためのコンピュータシミュレーションを実施した。振動回転状態の時間発展は、緊密結合法を用いて計算した。コムのスペクトル周波数をLiClの回転遷移周波数に一致させることにより、LiClとLiClに対して、LiClだけを高回転状態(J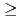 100)に励起することができた。使用したコムでは、振動状態励起は誘起されなかった。さらに、高回転状態のLiClの第二パルスによる解離過程を、波束法を用いて計算した。設定したパルスパラメータにより、振動回転状態(v=0, J=150)にあるLiClの約50%を解離することができた。これらの結果により、テラヘルツ領域の光学パルスを用いた二原子分子の同位体分離法の可能性を示した。
100)に励起することができた。使用したコムでは、振動状態励起は誘起されなかった。さらに、高回転状態のLiClの第二パルスによる解離過程を、波束法を用いて計算した。設定したパルスパラメータにより、振動回転状態(v=0, J=150)にあるLiClの約50%を解離することができた。これらの結果により、テラヘルツ領域の光学パルスを用いた二原子分子の同位体分離法の可能性を示した。
 )
)黒崎 譲; 横山 啓一
Computational and Theoretical Chemistry, 999, p.239 - 245, 2012/11
被引用回数:5 パーセンタイル:10.12(Chemistry, Physical)Potential energy curves (PECs) for the low-lying states of the diatomic cesium iodide cation (CsI) have been calculated using the internally-contracted MRSDCI method with relativistic pseudpotentials. First, we calculate PECs for spin-orbit (SO)-free 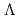 -S states,
-S states, 
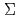 and
and 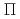 , stemming from the triply degenerate P state of I. Then we obtain PECs for SO-included
, stemming from the triply degenerate P state of I. Then we obtain PECs for SO-included 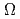 states, X3/2, 1/2(I), and 1/2(II), by diagonalizing the matrix of the electronic Hamiltonian plus SO coupling. It is found that all of the three state PECs, X3/2, 1/2(I), and 1/2(II), have shallow wells with the depths of 1.4-4.6 kcal mol
states, X3/2, 1/2(I), and 1/2(II), by diagonalizing the matrix of the electronic Hamiltonian plus SO coupling. It is found that all of the three state PECs, X3/2, 1/2(I), and 1/2(II), have shallow wells with the depths of 1.4-4.6 kcal mol . It is also predicted that transition dipole moments (TDMs) among these states have values of non-negligible amounts around equilibrium internuclear distances and the TDMs can contribute to the optical transition between the states.
. It is also predicted that transition dipole moments (TDMs) among these states have values of non-negligible amounts around equilibrium internuclear distances and the TDMs can contribute to the optical transition between the states.
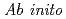 MRSDCI study on the low-lying electronic states of the lithium chloride molecule (LiCl)
MRSDCI study on the low-lying electronic states of the lithium chloride molecule (LiCl)黒崎 譲; 横山 啓一
Journal of Chemical Physics, 137(6), p.064305_1 - 064305_9, 2012/08
被引用回数:15 パーセンタイル:46.89(Chemistry, Physical)Potential energy curves (PECs) for the low-lying states of the lithium chloride molecule (LiCl) have been calculated using the MRSDCI method with the aug-cc-PV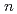 Z (AVZ) and aug-cc-PCVZ (ACVZ) basis sets, where = T, Q, and 5. First, we calculate PECs for 7 spin-orbit (SO)-free -S states, and then obtain PECs for 13 SO by diagonalizing the matrix of the electronic Hamiltonian plus the Breit-Pauli SO Hamiltonian. It is thus confirmed that to accurately predict the spectroscopic constants we need to include core-electron correlation in the CI expansion and use the basis sets designed to describe core-valence correlation, i.e., ACVZ.
Z (AVZ) and aug-cc-PCVZ (ACVZ) basis sets, where = T, Q, and 5. First, we calculate PECs for 7 spin-orbit (SO)-free -S states, and then obtain PECs for 13 SO by diagonalizing the matrix of the electronic Hamiltonian plus the Breit-Pauli SO Hamiltonian. It is thus confirmed that to accurately predict the spectroscopic constants we need to include core-electron correlation in the CI expansion and use the basis sets designed to describe core-valence correlation, i.e., ACVZ.
黒崎 譲; 市原 晃; 横山 啓一
Journal of Chemical Physics, 135(5), p.054103_1 - 054103_6, 2011/08
被引用回数:5 パーセンタイル:15.54(Chemistry, Physical)We have presented the OCT formulation to calculate optimal fields that can control the full ensemble of randomly-oriented molecules having different field-free Hamiltonians. The theory is applied to the fifty-fifty mixture of randomly-oriented CsI and CsI isotopomers and an optimal field is sought to achieve isotope-selective vibrational excitations with high efficiency. Rotational motion is frozen and two total times 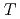 's of electric field duration, 460000 and 920000 au (11.1 and 22.2 ps), are chosen in the present calculation. As a result, the final yields for = 460000 and 920000 a.u. are calculated to be 0.706 and 0.815, respectively. The relatively high final yield obtained for = 920000 a.u. strongly suggests that a single laser pulse can control the full ensemble of randomly-oriented non-identical molecules. The result is quite encouraging in terms of the application to isotope-separation processes.
's of electric field duration, 460000 and 920000 au (11.1 and 22.2 ps), are chosen in the present calculation. As a result, the final yields for = 460000 and 920000 a.u. are calculated to be 0.706 and 0.815, respectively. The relatively high final yield obtained for = 920000 a.u. strongly suggests that a single laser pulse can control the full ensemble of randomly-oriented non-identical molecules. The result is quite encouraging in terms of the application to isotope-separation processes.
黒崎 譲; 横山 啓一; 横山 淳
Computational and Theoretical Chemistry, 963(2-3), p.245 - 255, 2011/02
被引用回数:6 パーセンタイル:13.16(Chemistry, Physical)We study, using quantum optimal control theory (OCT), isotope-selective vibrational excitations on the ground-state potential energy curve for the mixture of two pure ensembles for CsI and CsI. Initial states are set to the condition that both CsI and CsI are in the vibrational ground level, i.e., (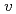
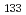 ,
, 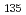 ) = (0, 0), and target states are set to three: (, ) = (0, 2), (0, 3), and (0, 4), where molecular orientations are fixed parallel to the field polarization vector. Three total times (i.e., pulse durations), T = 460,000 au, 920,000 au, and 1,840,000 au, are set in the calculations and nine cases for the combination of target state and total time are investigated. It is suggested from the computational results that even when T is short, high-yield transitions into energetically-separated target states are possible through excitations skipping over more than one vibrational level at a time with intense fields including high overtones.
) = (0, 0), and target states are set to three: (, ) = (0, 2), (0, 3), and (0, 4), where molecular orientations are fixed parallel to the field polarization vector. Three total times (i.e., pulse durations), T = 460,000 au, 920,000 au, and 1,840,000 au, are set in the calculations and nine cases for the combination of target state and total time are investigated. It is suggested from the computational results that even when T is short, high-yield transitions into energetically-separated target states are possible through excitations skipping over more than one vibrational level at a time with intense fields including high overtones.
板倉 隆二; 長谷川 宗良*; 黒崎 譲; 横山 淳; 大島 康裕*
Journal of Physical Chemistry A, 114(42), p.11202 - 11209, 2010/07
被引用回数:12 パーセンタイル:36.84(Chemistry, Physical)強レーザー場中の分子は、誘起双極子によって、非断熱回転励起する。レーザー強度が上がるとイオン化が起こり、回転励起とイオン化が同時進行する。イオン化には配向角度依存性があり、イオン化によって中性分子の回転波束が変化する。本研究は、フェムト秒強レーザーパルス照射後のNO分子の回転状態分布について、ナノ秒色素レーザーを用いた共鳴2光子イオン化回転スペクトル測定によって明らかにした。また、イオン化を取り込んだ時間依存シュレーディンガー方程式の数値計算を行い、実験結果と比較した結果、42TW/cm程度のレーザー強度では、分子軸がレーザー偏光方向に垂直なときに、イオン化確率が大きくなることが明らかとなった。
黒崎 譲; 横山 啓一; 横山 淳
Journal of Molecular Structure; THEOCHEM, 913(1-3), p.38 - 42, 2009/11
We demonstrate, using optimal control theory, that perfect isotope-selective molecular vibrational excitations on the ground-state potential curve in multilevel systems can be completed in much shorter time scales than those in two-level systems. We consider a gaseous isotopic mixture of cesium iodide (CsI and CsI) and, using two-level and multilevel systems, try to obtain electric fields that drive different isotopes into different vibrational levels. As a result, we find that in multilevel systems isotope-selective excitation processes can be controlled much faster, which we call multilevel effect. It is likely that this effect makes use of the large isotope shifts of higher vibrational levels than the lowest two.
黒崎 譲; 横山 啓一; 横山 淳
Journal of Chemical Physics, 131(14), p.144305_1 - 144305_9, 2009/10
被引用回数:9 パーセンタイル:28.12(Chemistry, Physical)Quantum optimal control calculations have been carried out for isotope-selective vibrational excitations of the cesium iodide (CsI) molecule. Considering a gaseous isotopic mixture of CsI and CsI, the initial state is set to the condition that both CsI and CsI are in the vibrational ground level ( = 0) and the target state is that CsI is in the = 0 level while CsI in the first-excited level ( = 1). We find, using the density-matrix formalism, that perfect isotope-selective excitations for multilevel systems including more than ten lowest vibrational states can be completed in much shorter time scales than those for two-level systems. This multilevel effect comes from the large isotope shifts of the vibrational levels of 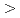 1. To check the reliability of the calculation we also employ the conventional wave-packet formalism and obtain almost the same results as those with the density-matrix formalism.
1. To check the reliability of the calculation we also employ the conventional wave-packet formalism and obtain almost the same results as those with the density-matrix formalism.
黒崎 譲; Artamonov, M.*; Ho, T.-S.*; Rabitz, H.*
Journal of Chemical Physics, 131(4), p.044306_1 - 044306_8, 2009/07
被引用回数:25 パーセンタイル:64.00(Chemistry, Physical)Quantum optimal control simulations have been carried out molecular isomerization dynamics of a one-dimensional (1D) reaction-path model involving a dominant competing dissociation channel. The 1D intrinsic reaction coordinate model mimics the ozone open cyclic ring isomerization along the minimum energy path that successively connects the ozone cyclic ring minimum, the transition state (TS), the open (global) minimum, and the dissociative O + O asymptote on the O ground-state 1A' potential energy surface. The molecular orientation of the modeled ozone is held constant with respect to the laser-field polarization and several optimal fields are found that all produce nearly perfect isomerization. The optimal control fields are characterized by distinctive high temporal peaks as well as low frequency components, thereby enabling abrupt transfer of the time-dependent wave packet over the TS from the open minimum to the targeted ring minimum.
ground-state 1A' potential energy surface. The molecular orientation of the modeled ozone is held constant with respect to the laser-field polarization and several optimal fields are found that all produce nearly perfect isomerization. The optimal control fields are characterized by distinctive high temporal peaks as well as low frequency components, thereby enabling abrupt transfer of the time-dependent wave packet over the TS from the open minimum to the targeted ring minimum.
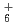 clusters; Direct molecular dynamics study
clusters; Direct molecular dynamics study黒崎 譲; 清水 裕太*; 熊谷 純*
Chemical Physics Letters, 455(1-3), p.59 - 63, 2008/03
被引用回数:2 パーセンタイル:5.01(Chemistry, Physical)Hクラスターとその同位体置換クラスター、[H(H)H], [H(H)D], [H(HD)H]について直接非経験的分子動力学計算を行った。計算の結果、[H(H)D]及び[H(HD)H]クラスターの平均構造は非対称的になるため、超微細結合定数(HFCC)を決定するスピン密度が偏ることが予測された。この予測は、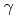 線照射された固体パラ水素中に生成するとされるこれらのクラスターのHFCCについての最近のESR観測の結果と一致している。よって今回の計算結果は、固体パラ水素中におけるHクラスターの生成をより強く裏付けるものである。
線照射された固体パラ水素中に生成するとされるこれらのクラスターのHFCCについての最近のESR観測の結果と一致している。よって今回の計算結果は、固体パラ水素中におけるHクラスターの生成をより強く裏付けるものである。
黒崎 譲
Journal of Molecular Structure; THEOCHEM, 850(1-3), p.9 - 16, 2008/02
被引用回数:12 パーセンタイル:29.10(Chemistry, Physical)最低励起三重項状態(T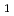 )のポテンシャルエネルギー面(PES)上でのアセトアルデヒドの光分解における水素原子生成チャンネル: CDCHOCDCHO+D(1); CDCHOCDCO+H(2)について、密度汎関数理論(DFT)に基づく直接分子動力学計算を実施した。DFT法はB3LYPで、基底関数はcc-pVDZを用いた。計算の結果、生成した水素原子の平均の運動エネルギーは反応1, 2でそれぞれ18.3, 16.6kcal molであった。これらは、実験値[Kang et al., Chem. Phys. Lett. 434 (2007) 6]の半分から2/3の値であった。計算値が実験値に比べてかなり小さい理由は、反応1, 2の生成物側から見た障壁がB3LYP/cc-pVDZレベルでは小さく見積もられていることにある。しかしながら、ここでの計算結果は実験値と定性的には一致しており、これらの反応がTPES上で起こることを支持するものである。
)のポテンシャルエネルギー面(PES)上でのアセトアルデヒドの光分解における水素原子生成チャンネル: CDCHOCDCHO+D(1); CDCHOCDCO+H(2)について、密度汎関数理論(DFT)に基づく直接分子動力学計算を実施した。DFT法はB3LYPで、基底関数はcc-pVDZを用いた。計算の結果、生成した水素原子の平均の運動エネルギーは反応1, 2でそれぞれ18.3, 16.6kcal molであった。これらは、実験値[Kang et al., Chem. Phys. Lett. 434 (2007) 6]の半分から2/3の値であった。計算値が実験値に比べてかなり小さい理由は、反応1, 2の生成物側から見た障壁がB3LYP/cc-pVDZレベルでは小さく見積もられていることにある。しかしながら、ここでの計算結果は実験値と定性的には一致しており、これらの反応がTPES上で起こることを支持するものである。

