


Direct density-functional (DFT) molecular-dynamics (MD) calculations have been carried out for the following two hydrogen atom production channels in acetaldehyde photodissociation on the lowest triplet-state (T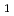 ) potential energy surface (PES): CD
) potential energy surface (PES): CD CHO
CHO CD
CD CHO+D(1); CDCHOCDCO+H(2). The employed DFT method was B3LYP with the cc-pVDZ basis set. The average product hydrogen kinetic energies estimated from the results of the direct DFT MD calculations were 18.3 and 16.6 kcal mol
CHO+D(1); CDCHOCDCO+H(2). The employed DFT method was B3LYP with the cc-pVDZ basis set. The average product hydrogen kinetic energies estimated from the results of the direct DFT MD calculations were 18.3 and 16.6 kcal mol for reactions 1 and 2, respectively, and these were half - two thirds of the previously measured values [Kang et al. Chem. Phys. Lett. 434 (2007) 6]. This is because of the low reverse barrier heights predicted at the B3LYP/cc-pVDZ level. The present results for the product hydrogen kinetic energies, however, agree qualitatively with the experimental measurements and strongly supports the mechanisms taking place on the T PES.
for reactions 1 and 2, respectively, and these were half - two thirds of the previously measured values [Kang et al. Chem. Phys. Lett. 434 (2007) 6]. This is because of the low reverse barrier heights predicted at the B3LYP/cc-pVDZ level. The present results for the product hydrogen kinetic energies, however, agree qualitatively with the experimental measurements and strongly supports the mechanisms taking place on the T PES.
Language |
: | English |
|---|---|---|
Journal |
: | |
Volume |
: | 850 |
Number |
: | 1-3 |
Pages |
: | p.9 - 16 |
Publication Year/Month |
: | 2008/02 |
Paper URL |
: |
|
Keywords |
: | no keyword |
Accesses |
: |
- Accesses |
|---|---|---|
InCites™ |
: |
Percentile:29.10 Category:Chemistry, Physical |
Altmetrics |
: |
[CLARIVATE ANALYTICS], [WEB OF SCIENCE], [HIGHLY CITED PAPER & CUP LOGO] and [HOT PAPER & FIRE LOGO] are trademarks of Clarivate Analytics, and/or its affiliated company or companies, and used herein by permission and/or license.
