


Nakazawa, Tetsuya; Igarashi, Takahiro; Tsuru, Tomohito; Kaji, Yoshiyuki
Fe, Ni、及びFe-Niクラスターについて密度汎関数法を基礎とした計算科学的手法により調べた。クラスターの構造はUB3LYP/LanL2DZレベルにおいて対称性の制約のもと最適化した。本研究では平衡構造と結合エネルギーを示し、密度解析の結果や電子配置とともに議論した。さらに、Fe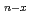 Ni
Ni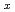 クラスターの結合エネルギーはFe原子のNi原子での連続的な置換によって緩やかに減少することがわかった。異性体間における全エネルギーの比較からFe-Niの混合クラスターではNi原子はエネルギー的にクラスタリングをしやすいことが明らかとなった。本研究は、照射した圧力容器で観察されているNiクラスターやステンレス鋼で見られる照射誘起偏析のNi富化などの実験結果と定性的に一致している。
クラスターの結合エネルギーはFe原子のNi原子での連続的な置換によって緩やかに減少することがわかった。異性体間における全エネルギーの比較からFe-Niの混合クラスターではNi原子はエネルギー的にクラスタリングをしやすいことが明らかとなった。本研究は、照射した圧力容器で観察されているNiクラスターやステンレス鋼で見られる照射誘起偏析のNi富化などの実験結果と定性的に一致している。
The clusters of Fe, Ni, and Fe-Ni are investigated computationally using a density functional approach. The geometries of clusters are optimized under the constraint of well-defined point group symmetries at the UB3LYP/LanL2DZ level. The equilibrium geometries and binding energies are presented and discussed, together with natural populations and natural electron configurations. In addition, the binding energies of FeNi clusters are found to generally decrease by successive substitutions of Ni atoms for Fe atoms. For Fe Ni clusters, the comparisons on total energies between isomers indicate that Ni atoms energetically prefer clustering in the mixed Fe-Ni clusters. The calculations for FeNi clusters show that the clustering leads to a segregation of Ni atoms from Fe atoms.
発表言語 |
: | English |
|---|---|---|
掲載資料名 |
: | |
巻 |
: | 46 |
号 |
: | 2 |
ページ数 |
: | p.367 - 375 |
発行年月 |
: | 2009/08 |
キーワード |
: | no keyword |
論文URL |
: |
|
|---|---|---|
論文解説記事 |
: |
Access |
: |
- Accesses |
|---|---|---|
InCites™ |
: |
パーセンタイル:59.31 分野:Materials Science, Multidisciplinary |
Altmetrics |
: |
[CLARIVATE ANALYTICS], [WEB OF SCIENCE], [HIGHLY CITED PAPER & CUP LOGO] and [HOT PAPER & FIRE LOGO] are trademarks of Clarivate Analytics, and/or its affiliated company or companies, and used herein by permission and/or license.


