


Nakazawa, Tetsuya; Igarashi, Takahiro 
 ; Tsuru, Tomohito
; Tsuru, Tomohito ![]() ; Kaji, Yoshiyuki
; Kaji, Yoshiyuki
The clusters of Fe, Ni, and Fe-Ni are investigated computationally using a density functional approach. The geometries of clusters are optimized under the constraint of well-defined point group symmetries at the UB3LYP/LanL2DZ level. The equilibrium geometries and binding energies are presented and discussed, together with natural populations and natural electron configurations. In addition, the binding energies of Fe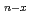 Ni
Ni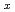 clusters are found to generally decrease by successive substitutions of Ni atoms for Fe atoms. For Fe Ni clusters, the comparisons on total energies between isomers indicate that Ni atoms energetically prefer clustering in the mixed Fe-Ni clusters. The calculations for FeNi clusters show that the clustering leads to a segregation of Ni atoms from Fe atoms.
clusters are found to generally decrease by successive substitutions of Ni atoms for Fe atoms. For Fe Ni clusters, the comparisons on total energies between isomers indicate that Ni atoms energetically prefer clustering in the mixed Fe-Ni clusters. The calculations for FeNi clusters show that the clustering leads to a segregation of Ni atoms from Fe atoms.
Language |
: | English |
|---|---|---|
Journal |
: | |
Volume |
: | 46 |
Number |
: | 2 |
Pages |
: | p.367 - 375 |
Publication Year/Month |
: | 2009/08 |
Paper URL |
: |
|
Keywords |
: | no keyword |
Accesses |
: |
- Accesses |
|---|---|---|
InCites™ |
: |
Percentile:58.09 Category:Materials Science, Multidisciplinary |
Altmetrics |
: |
[CLARIVATE ANALYTICS], [WEB OF SCIENCE], [HIGHLY CITED PAPER & CUP LOGO] and [HOT PAPER & FIRE LOGO] are trademarks of Clarivate Analytics, and/or its affiliated company or companies, and used herein by permission and/or license.
