


 H
H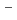 and FH
and FH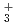
Suzuki, Kimichi*; Ishibashi, Hiroaki*; Yagi, Kiyoshi*; Shiga, Motoyuki 
![]()
 ; Tachikawa, Masanori*
; Tachikawa, Masanori*
The quantum nature of the strong hydrogen bonds for the FH and FH ions and their deuterated isotopomers at the room temperature has been studied using ab initio path integral molecular dynamics (PIMD) simulations. It is found that, for both of these ions, the hydrogen-bonded H/D atoms largely fluctuate around the central position of two F atoms. The average FH/FF distances of FH and FH are longer than the average FD/FF distances of F D
D and FH due to the primary/secondary isotope effects, which stem from the difference of the quantum nature of H and D nuclei. These results are compared with the family of Zundel-type ions, OH
and FH due to the primary/secondary isotope effects, which stem from the difference of the quantum nature of H and D nuclei. These results are compared with the family of Zundel-type ions, OH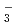 , NH
, NH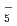 , OH
, OH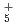 , and NH
, and NH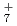 , which have been studied previously with the same ab initio PIMD approach. A comparison is also made with the previous experimental and ab initio vibrational configuration interaction results of FH.
, which have been studied previously with the same ab initio PIMD approach. A comparison is also made with the previous experimental and ab initio vibrational configuration interaction results of FH.
Language |
: | English |
|---|---|---|
Journal |
: | |
Volume |
: | 26 |
Number |
: | |
Pages |
: | p.207 - 216 |
Publication Year/Month |
: | 2012/08 |
Paper URL |
: |
|
Keywords |
: | no keyword |
Research Facility |
: |
Accesses |
: |
- Accesses |
|---|---|---|
Altmetrics |
: |
[CLARIVATE ANALYTICS], [WEB OF SCIENCE], [HIGHLY CITED PAPER & CUP LOGO] and [HOT PAPER & FIRE LOGO] are trademarks of Clarivate Analytics, and/or its affiliated company or companies, and used herein by permission and/or license.
