


 H
H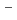 and FH
and FH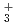 HとFHの第一原理経路積分分子動力学シミュレーション
HとFHの第一原理経路積分分子動力学シミュレーション鈴木 机倫*; 石橋 宏章*; 八木 清*; 志賀 基之 
![]()
 ; 立川 仁典*
; 立川 仁典*
Suzuki, Kimichi*; Ishibashi, Hiroaki*; Yagi, Kiyoshi*; Shiga, Motoyuki; Tachikawa, Masanori*
Zundel型イオンは特異的に強い水素結合を持つが、その詳細な構造は知られていない。本研究では、第一原理経路積分分子動力学シミュレーションを用いて、フッ素のZundel型イオンであるFHイオン, FHイオンとその重水素置換体の構造について調べた。これらのイオンの水素結合において、H/D原子は、原子核の量子性のため、F原子間の中心のまわりで大きく構造がゆらいでいることがわかった。また、FHやFHのFHやFF原子間距離は、その重水素置換体のFDやFDのものよりも長くなる。この結果について、他のZundel型イオンOH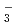 , NH
, NH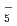 , OH
, OH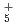 , NH
, NH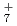 と比較したところ、それらの重原子間の平均距離とその重水素置換効果に相関があることを発見した。なお、FHについては、実験結果や、振動配置間相互作用法による計算結果とよく一致していることがわかり、用いた手法の精度が極めて高いことが立証できた。本成果は、水素結合等での同位体効果やその量子性などを考慮する第一原理計算手法の開発とその検証にあたり、さまざまな分野での応用が期待できる。
と比較したところ、それらの重原子間の平均距離とその重水素置換効果に相関があることを発見した。なお、FHについては、実験結果や、振動配置間相互作用法による計算結果とよく一致していることがわかり、用いた手法の精度が極めて高いことが立証できた。本成果は、水素結合等での同位体効果やその量子性などを考慮する第一原理計算手法の開発とその検証にあたり、さまざまな分野での応用が期待できる。
The quantum nature of the strong hydrogen bonds for the FH and FH ions and their deuterated isotopomers at the room temperature has been studied using ab initio path integral molecular dynamics (PIMD) simulations. It is found that, for both of these ions, the hydrogen-bonded H/D atoms largely fluctuate around the central position of two F atoms. The average FH/FF distances of FH and FH are longer than the average FD/FF distances of F D
D and FH due to the primary/secondary isotope effects, which stem from the difference of the quantum nature of H and D nuclei. These results are compared with the family of Zundel-type ions, OH, NH, OH, and NH, which have been studied previously with the same ab initio PIMD approach. A comparison is also made with the previous experimental and ab initio vibrational configuration interaction results of FH.
and FH due to the primary/secondary isotope effects, which stem from the difference of the quantum nature of H and D nuclei. These results are compared with the family of Zundel-type ions, OH, NH, OH, and NH, which have been studied previously with the same ab initio PIMD approach. A comparison is also made with the previous experimental and ab initio vibrational configuration interaction results of FH.
発表言語 |
: | English |
|---|---|---|
掲載資料名 |
: | |
巻 |
: | 26 |
号 |
: | |
ページ数 |
: | p.207 - 216 |
発行年月 |
: | 2012/08 |
キーワード |
: | no keyword |
論文URL |
: |
|
|---|---|---|
使用施設 |
: | |
論文解説 |
: |
Access |
: |
- Accesses |
|---|---|---|
Altmetrics |
: |
[CLARIVATE ANALYTICS], [WEB OF SCIENCE], [HIGHLY CITED PAPER & CUP LOGO] and [HOT PAPER & FIRE LOGO] are trademarks of Clarivate Analytics, and/or its affiliated company or companies, and used herein by permission and/or license.
