


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
志賀 基之; Thomsen, B.; 君塚 肇*
Physical Review B, 109(5), p.054303_1 - 054303_12, 2024/02
パラジウム中の水素の中性子非弾性散乱スペクトルを有限温度での核量子効果を考慮して計算した。経路積分に基づく半古典分子動力学と機械学習ポテンシャルを組合せた計算手法を用いた。計算されたスペクトルは、水素原子の振動励起の基本波と第一高調波に対応するピークの位置と強度に関して、実験スペクトルとよく一致した。古典分子動力学法との比較から、中性子非弾性散乱スペクトルにおいて核量子効果が本質的な役割を果たしていることが示された。
志賀 基之; Thomsen, B.; 永井 佑紀
アンサンブル, 25(4), p.303 - 310, 2023/10
並列分子シミュレーションソフトウェア「PIMD」を紹介する。第一原理経路積分分子動力学による水の構造、リングポリマー分子動力学による金属中水素の量子拡散、超伝導体の機械学習ポテンシャル作成とフォノン物性、メタダイナミクスによる多価アルコール脱水反応などの具体例を通じて、その使用法を解説する。
Kwon, H.*; 志賀 基之; 君塚 肇*; 小田 卓司*
Acta Materialia, 247, p.118739_1 - 118739_11, 2023/04
被引用回数:3 パーセンタイル:74.65(Materials Science, Multidisciplinary)機械学習によるモーメントテンソルポテンシャルを用いた経路積分シミュレーションから、体心立方格子金属(Nb, Fe, W)中の希薄水素の拡散係数を密度汎関数理論レベルの精度で推定した。この計算結果は、精度が高いと考えられるいくつかの実験結果と大いに一致した。また、実験結果と矛盾なく同位体効果を再現した。
志賀 基之
Journal of Computational Chemistry, 43(27), p.1864 - 1879, 2022/10
被引用回数:3 パーセンタイル:42.81(Chemistry, Multidisciplinary)経路積分法に基づいて、量子振動動力学のための新たな近似法(Brownian chain molecular dynamics: BCMD)を提案する。セントロイド分子動力学法やリングポリマー動力学法などの従来法では、量子振動スペクトルが低温条件で正しく計算できない。そこで、本研究では、その問題の源である非物理的な共鳴や振動回転カップリングを抑制するため、原子の非重心自由度に対して過減衰ランジュバン方程式を導入する。軽水や重水などの赤外吸収スペクトルへの応用例を通じて、この近似を検証する。また、BCMD法を電子状態理論と組み合わせた第一原理シミュレーションを実証する。
君塚 肇*; Thomsen, B.; 志賀 基之
Journal of Physics; Energy (Internet), 4(3), p.034004_1 - 034004_13, 2022/07
被引用回数:8 パーセンタイル:75.5(Energy & Fuels)パラジウム-水素系の人工ニューラルネットワークに基づく原子間ポテンシャルを開発するとともに、パラジウム結晶中の水素同位体の量子拡散について、経路積分分子動力学計算を行った。50-1500Kの広い温度範囲で軽水素と重水素の拡散係数を計算し、低温と高温における拡散機構の違いを明らかにした。
 path integral simulations
path integral simulationsThomsen, B.; 志賀 基之
Physical Chemistry Chemical Physics, 24(18), p.10851 - 10859, 2022/05
被引用回数:2 パーセンタイル:16.81(Chemistry, Physical)The heavy hydrogen isotopes D and T are found in trace amounts in water, and they can when their concentration rises play an intricate role in modulating the physical properties of the liquid. We present an analysis of the microscopic structures of ambient light water (H O(l)), heavy water(DO(l)), TO(l), HDO(aq) and HTO(aq) studied by path integral molecular dynamics (PIMD). Unlike previous PIMD investigations of HO(l) and DO(l) [Chen et al., Phys. Rev. Lett., 2003, 91, 215503] [Machida et al., J. Chem. Phys., 2017, 148, 102324] we do find that DO(l) is more structured than HO(l), as is predicted by experiment. The agreement between experiment and our simulation for HO(l) and DO(l) allows us to accurately predict intra- and intermolecular structures of TO(l) HDO(aq) and HTO(aq). TO(l) is found to have a similar intermolecular structure to that of DO(l), while the intramolecular structure is more compact, giving rise to a smaller dipole moment that that if HO(l) and DO(l). For the mixed isotope species, HDO(aq) and HTO(aq), we find smaller dipole moments and less hydrogen bonds when compared with the pure species HO and DO. We can attribute this effect to the relative compactness of the mixed isotope species, which results in a lower dipole moment when compared to that of the pure species.
O(l)), heavy water(DO(l)), TO(l), HDO(aq) and HTO(aq) studied by path integral molecular dynamics (PIMD). Unlike previous PIMD investigations of HO(l) and DO(l) [Chen et al., Phys. Rev. Lett., 2003, 91, 215503] [Machida et al., J. Chem. Phys., 2017, 148, 102324] we do find that DO(l) is more structured than HO(l), as is predicted by experiment. The agreement between experiment and our simulation for HO(l) and DO(l) allows us to accurately predict intra- and intermolecular structures of TO(l) HDO(aq) and HTO(aq). TO(l) is found to have a similar intermolecular structure to that of DO(l), while the intramolecular structure is more compact, giving rise to a smaller dipole moment that that if HO(l) and DO(l). For the mixed isotope species, HDO(aq) and HTO(aq), we find smaller dipole moments and less hydrogen bonds when compared with the pure species HO and DO. We can attribute this effect to the relative compactness of the mixed isotope species, which results in a lower dipole moment when compared to that of the pure species.
赤沢 第輔; 佐々木 岳彦*; 長坂 将成*; 志賀 基之
Journal of Chemical Physics, 156(4), p.044202_1 - 044202_7, 2022/01
被引用回数:3 パーセンタイル:50.03(Chemistry, Physical)セルロースの水和構造は、分子レベルでのバイオマスセルロースの加水分解メカニズムの理解のために非常に重要である。本論文では、セルロースの2糖の単量体であるセロビオースの水溶液について、そのX線吸収分光法(XAS)スペクトルの測定と理論計算を組み合わせた研究について報告する。測定に関する項目では高解像度の炭素K端のXASスペクトルの測定について報告する。理論計算の項目では、第一原理分子動力学計算と励起状態計算を用いてXASのシミュレーションを求める手法と結果ついて論ずる。セロビオースのXASには289.3eV, 290.7eV, 293.6eVの3つのピークがあり、それぞれアルコール基,ヘミアセタール基,アルコール及びヘミアセタール基の両方に対応することがわかった。さらに、25 Cから60Cまでの温度変化に対してスペクトルの高さが規則的に変化し、それがセロビオースと水分子の水素結合の数の変化を表していることがわかった。このスペクトルの変化は様々な環境におけるセルロースの水和構造を知るための情報として有用であると考えられる。
Cから60Cまでの温度変化に対してスペクトルの高さが規則的に変化し、それがセロビオースと水分子の水素結合の数の変化を表していることがわかった。このスペクトルの変化は様々な環境におけるセルロースの水和構造を知るための情報として有用であると考えられる。
 study of nuclear quantum effects on sub- and supercritical water
study of nuclear quantum effects on sub- and supercritical waterThomsen, B.; 志賀 基之
Journal of Chemical Physics, 155(19), p.194107_1 - 194107_11, 2021/11
被引用回数:6 パーセンタイル:52.59(Chemistry, Physical)The structures of water in the ambient, sub-, and supercritical conditions at various densities were studied systematically by path integral molecular dynamics simulations. It was found that the nuclear quantum effects (NQEs) have significant impact on the structure of hydrogen bonds in close contact, not only in the ambient condition but also in the sub-, and supercritical conditions. The NQEs on the structure beyond the hydrogen bond contact are important in ambient water, but not much for water in the sub-, and supercritical conditions. The NQEs are furthermore important for determining the number of hydrogen bonds in the ambient conditions, this role is however diminished in the sub- and supercritical conditions. The NQEs does nevertheless show their importance in determining the intramolecular structure of water and the close contact structures of the hydrogen bonds, even at sub- and supercritical conditions. Using the RPBE-D3 functional, the computed radial distribution functions for the ambient water are in excellent agreement with experimental data, upgrading our previous results using the BLYP-D2 functional [Machida, et al., J. Chem. Phys. 148, 102324 (2018)]. The computed radial distribution functions for water in the sub- and supercritical conditions were carefully compared with experiment. In particular, we found that the first peak in hydrogen pair distribution functions matches only when the NQEs are taken into account.
Pal, A.*; Pal, S.*; Verma, S.*; 志賀 基之; Nair, N. N.*
Journal of Computational Chemistry, 42(28), p.1996 - 2003, 2021/10
被引用回数:8 パーセンタイル:55.52(Chemistry, Multidisciplinary)温度加速断片サンプリングは、高次元の自由エネルギー地形の計算法である。従来法では、重み付きヒストグラム解析法を用いて計算の後処理を行うが必要があった。この研究では、平均力を使用して後処理を行わずに済む洗練された手法を確立する。これを用いると、アラニンジペプチドとトリペプチドの2次元および4次元の自由エネルギー地形が約kcal/molの誤差内で計算されること実証した。この方法は、計算化学における自由エネルギー計算において幅広い応用が期待される。
近藤 友美; 佐々木 岳彦*; 志賀 基之
Journal of Computational Chemistry, 42(25), p.1783 - 1791, 2021/09
被引用回数:1 パーセンタイル:6.77(Chemistry, Multidisciplinary)高温水溶液中の糖アルコール脱水反応は、有機溶媒を使用せずに環境に優しいバイオマス変換を可能にする重要な反応である。本研究では、高温酸性水中でのソルビトール(SBT)の脱水反応のメカニズムを理解するため、第一原理および半経験的電子状態理論に基づくメタダイナミクス(MTD)シミュレーションによる自由エネルギー解析を行った。反応機構は2分子求核置換型(S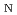 2)であり、それによって中間体として生成されたヒドロキシル基のプロトン化の自由エネルギーが酸性種の影響を受けることがわかった。また、5員エーテル生成物につながる反応経路の自由エネルギー障壁は、6員エーテル生成物につながるものよりも低いことがわかった。
2)であり、それによって中間体として生成されたヒドロキシル基のプロトン化の自由エネルギーが酸性種の影響を受けることがわかった。また、5員エーテル生成物につながる反応経路の自由エネルギー障壁は、6員エーテル生成物につながるものよりも低いことがわかった。
小林 恵太; 永井 佑紀; 板倉 充洋; 志賀 基之
Journal of Chemical Physics, 155(3), p.034106_1 - 034106_9, 2021/07
被引用回数:5 パーセンタイル:44.89(Chemistry, Physical)自己学習ハイブリッドモンテカルロ法は機械学習力場を利用することにより、第一原理計算の高速化を可能にする手法である。今回、自己学習ハイブリッドモンテカルロ法をNPTアンサンブルに適用し、液体シリカの解析を行った。本論文では自己学習ハイブリッドモンテカルロ法を用いることにより、厳密に第一原理計算の精度を保ちながら、高速に液体シリカの配置空間のサンプリングが可能であることを示した。また、液体シリカの構造因子の計算を行い実験データとの比較を行ったところ、両者はよい一致を見せた。
path integral simulations君塚 肇*; 志賀 基之
Physical Review Materials (Internet), 5(6), p.065406_1 - 065406_9, 2021/06
被引用回数:10 パーセンタイル:61.54(Materials Science, Multidisciplinary)金属内部における水素の動的挙動において、核量子効果は無視できない因子である。本研究では、核量子効果を考慮した第一原理積分分子動力学シミュレーションを用いて、面心立方金属Al, Ag, Cuにおける水素拡散を調べた。その結果、AgとCuにおける水素拡散の温度依存性は逆S字型の曲線となるのに対して、Alにおける水素拡散の温度依存性はC字型となることがわかった。この違いは、水素が最安定となる位置がAg, Cuでは八面体サイト、Alでは四面体サイトであることに由来する。このことから、水素拡散の核量子効果(零点振動とトンネリング)は安定サイトの異なる金属によって質的に異なることがわかった。
path integral molecular dynamicsThomsen, B.; 志賀 基之
Journal of Chemical Physics, 154(8), p.084117_1 - 084117_10, 2021/02
被引用回数:8 パーセンタイル:64.31(Chemistry, Physical)第一原理経路積分分子動力学シミュレーションにより、室温条件での水の同位体置換体の酸性度定数に対する核量子効果を計算した。この手法は、実験的に測定された液体D Oの酸性度定数を再現するだけでなく、希少性のために不明な液体TOや、HDOおよびHTO水溶液の酸性度定数の理論的予測も可能にする。計算結果は、核量子効果が酸性度定数の絶対的評価において不可欠な役割を果たすことを示している。
Oの酸性度定数を再現するだけでなく、希少性のために不明な液体TOや、HDOおよびHTO水溶液の酸性度定数の理論的予測も可能にする。計算結果は、核量子効果が酸性度定数の絶対的評価において不可欠な役割を果たすことを示している。
近藤 友美; 佐々木 岳彦*; Ruiz-Barragan, S.*; Ribas-Ari o, J.*; 志賀 基之; Ruiz-Barragan, S.*
o, J.*; 志賀 基之; Ruiz-Barragan, S.*
Journal of Computational Chemistry, 42(3), p.156 - 165, 2021/01
被引用回数:4 パーセンタイル:21.18(Chemistry, Multidisciplinary)本研究では、固定されたバイアスポテンシャル下の正準サンプリングによって、メタダイナミクスを改良する新たな計算手法を提案した。この手法は、2つ以上の自由エネルギー障壁を異なる条件または競合する条件での化学反応間で比較する場合などに役立つ。この方法を用いて、高温水溶液中の多価アルコール脱水反応の酸依存性を調べた。その結果、酸の種類によらず、この反応はS2メカニズムを介して進行することがわかった。一方、中間体として生成されるヒドロキシル基のプロトン化における自由エネルギー変化が、酸の種類によって影響を大きく受けることがわかった。
野口 良史*; 樋山 みやび*; 志賀 基之; 秋山 英文*; 杉野 修*
Journal of Chemical Physics, 153(20), p.201103_1 - 201103_6, 2020/11
被引用回数:2 パーセンタイル:11.43(Chemistry, Physical)第一原理分子動力学シミュレーションを用いて、第一励起状態のオキシルシフェリン水溶液の3つの異性体の安定化メカニズムを調べた。フェノラート-ケト異性体のみが励起状態で水分子に引き付けられ、近くの水分子との水素結合の数を増やすことによって安定化された。励起状態で最も安定な異性体はフェノラート-ケトであり、フェノラート-エノールおよびフェノール-エノラート異性体は、フェノラート-ケトよりもエネルギーがそれぞれ0.38eVおよび0.57eV高かった。この結果は、フェノラート-エノールが最も安定な異性体である基底状態の場合とは対照的である。
君塚 肇*; 尾方 成信*; 志賀 基之
日本物理学会誌, 75(8), p.484 - 490, 2020/08
水素は量子性を示す元素で、他の不純物原子にはない特異な拡散挙動を示す。本研究では、経路積分シミュレーションと電子状態計算を連成させた第一原理手法を用いて、パラジウム結晶中の水素同位体の拡散挙動を幅広い温度で予測した。その結果、高温領域では、温度の低下とともに量子ゆらぎの影響が顕在化することによって拡散抑制が生じ、アレニウスプロットが上に凸の折れ曲がることが示された。一方、低温域では、量子トンネル効果の発現によって拡散障壁は減少に転じ、アレニウスプロットは下に凸の折れ曲がることが示された。異なる温度領域における量子効果の競合は、水素拡散の特異な同位体効果を明瞭に説明する。
永井 佑紀; 奥村 雅彦; 小林 恵太*; 志賀 基之
Physical Review B, 102(4), p.041124_1 - 041124_6, 2020/07
被引用回数:13 パーセンタイル:66.36(Materials Science, Multidisciplinary)第一原理計算で得られたポテンシャルを再現するようなニューラルネットワーク(ANN)を構築して分子動力学を実行するのが機械学習分子動力学法である。ANNを構築する際の最適なトレーニングデータは、元々の第一原理分子動力学法で生成される原子配置とそのポテンシャルである。通常は、様々な原子配置とそのポテンシャルデータを大量に作成することで、目的の機械学習分子動力学法と同じようなポテンシャルを生成するANNを構築している。しかしながら、構築されたANNが元々の第一原理計算のポテンシャルを再現するという保証はない。さらに、4元素以上で構成されるような系の場合には、長時間の機械学習分子動力学法では計算が不安定になることがあり、機械学習分子動力学法の計算の精度や妥当性については常に慎重な議論が必要であった。本論文発表では、自己学習モンテカルロ法のアイディアを用いることで、得られた結果が統計的に厳密にオリジナルの第一原理計算分子動力学法の計算結果と等しい手法を開発したことを報告する。
Yan, L.*; 山本 良幸*; 志賀 基之; 杉野 修*
Physical Review B, 101(16), p.165414_1 - 165414_9, 2020/04
被引用回数:11 パーセンタイル:60.64(Materials Science, Multidisciplinary)電気化学において関心の高い、白金(111)表面上の高被覆率水素に対して、第一原理経路積分分子動力学シミュレーションを行った。その結果、室温において、量子効果と多体効果がともに重要な役割を果たすことを見出した。完全被覆率の場合、水素原子は、その強い反発力のため、アトップサイトまたは面心立方サイトに存在する。これに対して、被覆率2/3の場合には、水素原子は非局在化して、面心立方サイトと最密六方サイトに共存する。この結果は、白金表面への水素の被覆率依存性に関する長年の論争を解決する手がかりとして、量子多体効果が大変重要であることを示している。
君塚 肇*; 尾方 成信*; 志賀 基之
Physical Review B, 100(2), p.024104_1 - 024104_9, 2019/07
被引用回数:16 パーセンタイル:63.96(Materials Science, Multidisciplinary)結晶中の水素同位体の振る舞いは材料物理学における基本的なテーマである。広い温度範囲にわたる同位体拡散挙動は重要な知見だが、まだ完全な調査には至っていない。本研究では、電子と原子核の両方を量子力学的に扱った最先端の第一原理計算を行い、面心立方Pd金属中の水素同位体拡散係数の温度依存性を調べたところ、逆S型形状のアレニウスプロットを示すことがわかった。この異常な振る舞いは、温度・質量依存性の異なる核量子効果が競合することに起因する。その結果、80-400Kの限られた温度範囲で、三重水素(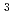 H)が軽水素(
H)が軽水素(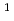 H)よりも速く拡散するという特性につながる。このメカニズムは、高温および低温での実験でそれぞれ観察されている正・逆同位体効果間がクロスオーバーを示す理論的根拠となることがわかった。
H)よりも速く拡散するという特性につながる。このメカニズムは、高温および低温での実験でそれぞれ観察されている正・逆同位体効果間がクロスオーバーを示す理論的根拠となることがわかった。
Chang, Y. L.*; 佐々木 岳彦*; Ribas-Ario, J.*; 町田 昌彦; 志賀 基之
Journal of Physical Chemistry B, 123(7), p.1662 - 1671, 2019/02
被引用回数:4 パーセンタイル:11.6(Chemistry, Physical)バイオマス由来ポリアルコールの脱水は、有機溶媒または金属触媒を使用せずに、高温水または高温炭酸水で制御可能な選択的反応の典型としてグリーンケミストリーにおいて最近注目を集めている。この研究では、酸性の高温水中での1,2,5-ペンタントリオールの競合分子内脱水反応のメカニズムを理解するために、第一原理メタダイナミクスとブルームーンアンサンブルシミュレーションに基づく自由エネルギー解析を行った。その結果、最も支配的なメカニズムは、水によるヒドロキシル基のプロトン化およびC-O結合の切断および形成が同時に起こる、プロトン支援S2型のプロセスであることがわかった。シミュレーションから見いだされた詳細なメカニズムは、どのようにして反応経路が熱水中で選択的であるか、そしてなぜ反応速度が酸性環境において加速されるかを示し、競合する多価アルコールの脱水プロセスについて実験結果の明確な説明を与える。
