


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
 @C
@C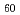 ; Photoelectron spectroscopy of the Li@C[PF
; Photoelectron spectroscopy of the Li@C[PF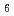
 ] salt and STM of the single Li@C molecules on Cu(111)
] salt and STM of the single Li@C molecules on Cu(111)山田 洋一*; Kuklin, A. V.*; 佐藤 翔*; 江坂 文孝; 角 直也*; Zhang, C.*; 佐々木 正洋*; Kwon, E.*; 笠間 泰彦*; Avramov, P. V.*; et al.
Carbon, 133, p.23 - 30, 2018/07
被引用回数:18 パーセンタイル:51.41(Chemistry, Physical)本研究では、超高真空中で高純度Li@C[PF]塩の蒸発によってLiイオン内包フラーレンを調製し、走査型トンネル顕微鏡(STM)により明瞭に観察することに成功した。また、STM観察に先立って、光電子分光およびX線吸収分光などにより測定したところ、Liは正、PFは負のチャージを帯びており、Cは中性であることが明らかとなった。
圓谷 志郎; Antipina, L. Y.*; Avramov, P.*; 大伴 真名歩*; 松本 吉弘*; 平尾 法恵; 下山 巖; 楢本 洋*; 馬場 祐治; Sorokin, P. B.*; et al.
Nano Research, 8(5), p.1535 - 1545, 2015/05
被引用回数:28 パーセンタイル:71.13(Chemistry, Physical)Direct growth of graphene on insulators is expected to yield significant improvements in performance of graphene-based electronic and spintronic devices. In this study, we successfully reveal atomic arrangement and electronic properties of the coherent heterostructure of single-layer graphene and  -Al
-Al O
O (0001). In the atomic arrangement analysis of single-layer graphene on -AlO(0001), we observed apparently contradicting results. The in-plane analysis shows that single-layer graphene grows not in the single-crystalline epitaxial manner but in the polycrystalline form with two strongly pronounced preferred orientations. This suggests the relatively weak interfacial interactions to be operative. But, we demonstrate that there exists unusually strong physical interactions between graphene and -AlO(0001), as evidenced by the short vertical distance between graphene and -AlO(0001) surface. The interfacial interactions are shown to be dominated by the electrostatic force involved in the graphene
(0001). In the atomic arrangement analysis of single-layer graphene on -AlO(0001), we observed apparently contradicting results. The in-plane analysis shows that single-layer graphene grows not in the single-crystalline epitaxial manner but in the polycrystalline form with two strongly pronounced preferred orientations. This suggests the relatively weak interfacial interactions to be operative. But, we demonstrate that there exists unusually strong physical interactions between graphene and -AlO(0001), as evidenced by the short vertical distance between graphene and -AlO(0001) surface. The interfacial interactions are shown to be dominated by the electrostatic force involved in the graphene 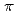 -system and the unsaturated electrons of the topmost O layer of -AlO(0001) rather than the van der Waals interactions. Such feature causes hole doping into graphene, which gives graphene a chance to slide on the -AlO(0001) surface with a small energy barrier despite the strong interfacial interactions.
-system and the unsaturated electrons of the topmost O layer of -AlO(0001) rather than the van der Waals interactions. Such feature causes hole doping into graphene, which gives graphene a chance to slide on the -AlO(0001) surface with a small energy barrier despite the strong interfacial interactions.
Kuzubov, A. A.*; Kovaleva, E. A.*; Avramov, P. V.*; Kuklin, A. V.*; Mikhaleva, N. S.*; Tomilin, F. N.*; 境 誠司; 圓谷 志郎; 松本 吉弘*; 楢本 洋*
Journal of Applied Physics, 116(8), p.084309_1 - 084309_4, 2014/08
被引用回数:9 パーセンタイル:36.08(Physics, Applied)The interaction between carbon and BN nanotubes (NT) and transition metal Co and Ni supports was studied using electronic structure calculations. Several configurations of interfaces were considered, and the most stable ones were used for electronic structure analysis. All NT/Co interfaces were found to be more energetically favorable than NT/Ni, and conductive carbon nanotubes demonstrates lightly stronger bonding than semiconducting ones. The presence of contact-induced spin polarization was established for all nanocomposites. It was found that the contact-induced polarization of BNNT leads to the appearance of local conductivity in the vicinity of the interface while the rest of the nanotube lattice remains to be insulating.
Avramov, P. V.*; Kuzubov, A. A.*; 境 誠司; 大伴 真名歩*; 圓谷 志郎; 松本 吉弘*; Eleseeva, N. S.*; Pomogaev, V. A.*; 楢本 洋*
Journal of Porphyrins and Phthalocyanines, 18(7), p.552 - 568, 2014/07
被引用回数:1 パーセンタイル:4.96(Chemistry, Multidisciplinary)A wide variety of planar and curved fused porphyrin/metalloporphyrin nanoclusters have been studied at the LC-GGA DFT level. It was found that curved and hollow-caged 0D and 1D nanoclusters are metastable and bear unique atomic and electronic structure and mechanical properties. Under different types of mechanical loads the nanoclusters display ultrastrong and superelastic properties. The curvature of the hollow cage nanoclusters leads to the redistribution of the metal d states near the Fermi level. The extremely high spin states allow one to use Fe-porphyrin nanoclusters as molecular supermagnets and logic quantum gates for holonomic quantum computations.
大伴 真名歩; 山内 泰*; Kuzubov, A. A.*; Eliseeva, N. S.*; Avramov, P.*; 圓谷 志郎; 松本 吉弘; 楢本 洋*; 境 誠司
Applied Physics Letters, 104(5), p.051604_1 - 051604_4, 2014/02
被引用回数:12 パーセンタイル:44.87(Physics, Applied)六方晶窒化ホウ素(h-BN)はグラフェン・スピントロニクスのトンネルバリア材料として有望視されている。本研究ではスピン偏極準安定ヘリウム脱励起分光法(SPMDS)を用いて、Ni(111)上の単層h-BNのスピン分解バンド構造を調べた。SPMDSの最表面敏感性により、Ni 3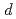 ピークの重複を受けずに部分的に占有されたギャップ内順位を検出できた。さらにこのギャップ内順位は大きなスピン偏極を持ち、Niの多数スピン側に偏極していることが示された。この正のスピン偏極は、h-BN/Ni(111)界面における-軌道混成によるものであると帰属できた。
ピークの重複を受けずに部分的に占有されたギャップ内順位を検出できた。さらにこのギャップ内順位は大きなスピン偏極を持ち、Niの多数スピン側に偏極していることが示された。この正のスピン偏極は、h-BN/Ni(111)界面における-軌道混成によるものであると帰属できた。
松本 吉弘; 圓谷 志郎; 小出 明広*; 大伴 真名歩; Avramov, P.; 楢本 洋*; 雨宮 健太*; 藤川 高志*; 境 誠司
Journal of Materials Chemistry C, 1(35), p.5533 - 5537, 2013/09
被引用回数:34 パーセンタイル:76.08(Materials Science, Multidisciplinary)The spin-electronic structures across the interface between single-layer graphene and a Ni(111) thin film are explored by employing the depth-resolved X-ray absorption and magnetic circular dichroism spectroscopy with the atomic layer resolution. The depth-resolved Ni L-edge analysis clarifies that the Ni atomic layers adjacent to the interface show a transition of the spin orientation to the perpendicular one in contrast with the in-plane one in the bulk region. The C K-edge analysis reveals the intensifying of the spin-orbital interactions induced by the 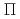 -d hybridization at the interface as well as out-of-plane spin polarization at the band region of graphene. The present study indicates the importance of the interface design at the atomic layer level for graphene-based spintronics.
-d hybridization at the interface as well as out-of-plane spin polarization at the band region of graphene. The present study indicates the importance of the interface design at the atomic layer level for graphene-based spintronics.
interface in the Fe-doped C film境 誠司; 松本 吉弘; 大伴 真名歩; 圓谷 志郎; Avramov, P.; Sorokin, P. B.*; 楢本 洋*
Synthetic Metals, 173, p.22 - 25, 2013/06
被引用回数:3 パーセンタイル:14.29(Materials Science, Multidisciplinary)A process of tunneling conduction and the spin-dependent resistivity change (so-called tunneling magnetoresitance effect) in the Fe-doped C film with a granular structure is investigated for the current-into-plane device. Cooperative tunneling (cotunneling) through several Fe nanoparticles is suggested to be operative at temperatures lower than 20 K. By considering the effect of cotunneling on the magnetoresistance ratio, it is successfully shown that the spin polarization of tunneling electrons generated at the Fe/C interface is much higher than that in Fe crystal at low temperature in a similar fashion to that at the Co/C interface in the Co-doped C films. A strong temperature dependence of spin polarization is observed, suggesting possible influences by the thermally-induced disorders ascribed in the Fe atoms bonded with C in the C-Fe compound.
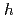 -BN/Ni nanocomposites
-BN/Ni nanocompositesAvramov, P.; Kuzubov, A. A.*; 境 誠司; 大伴 真名歩; 圓谷 志郎; 松本 吉弘; 楢本 洋*; Eliseeva, N. S.*
Journal of Applied Physics, 112(11), p.114303_1 - 114303_10, 2012/12
被引用回数:18 パーセンタイル:57.64(Physics, Applied)Atomic and electronic structure of graphene/Ni(111), -BN/Ni(111) and graphene/-BN/Ni(111) nanocomposites with different numbers of graphene and -BN layers and in different mutual arrangements of graphene/Ni and -BN/Ni at the interfaces was studied using LDA/PBC/PW technique. For the sake of comparison, corresponding graphene, -BN and graphene/-BN structures without the Ni plate were calculated using the same technique. It was suggested that C-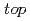 :C-
:C-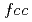 and N-:B- configurations are energetically favorable for the graphene/Ni and -BN/Ni interfaces, respectively. The Ni plate was found to induce a significant degree of spin polarization in graphene and -BN caused by direct exchange interactions of the electronic states located on different fragments.
and N-:B- configurations are energetically favorable for the graphene/Ni and -BN/Ni interfaces, respectively. The Ni plate was found to induce a significant degree of spin polarization in graphene and -BN caused by direct exchange interactions of the electronic states located on different fragments.
Avramov, P.; Fedorov, D. G.*; Sorokin, P. B.*; 境 誠司; 圓谷 志郎; 大伴 真名歩; 松本 吉弘; 楢本 洋*
Journal of Physical Chemistry Letters (Internet), 3(15), p.2003 - 2008, 2012/08
被引用回数:38 パーセンタイル:79.31(Chemistry, Physical)The atomic and electronic structure of narrow zigzag nanoribbons with finite length, consisting of graphene terminated by fluorine on one side, hexagonal ()-BN, and -SiC were studied with density functional theory. It is found that the asymmetry of nanoribbon edges causes a uniform curvature of the ribbons due to structural stress in the aromatic ring plane. Narrow graphene nanoribbons terminated with fluorine on one side demonstrate a considerable out-of-plane bend, suggesting that the nanoribbon is a fraction of a conical surface. It is shown that the intrinsic curvature of the narrow nanoribbons destroys the periodicity and results in a systematic cancellation of the dipole moment. The in- and out-of-plane curvature of thin arcs allows their closure in nanorings or cone fragments of giant diameter.
Antipina, L. Y.*; Avramov, P.; 境 誠司; 楢本 洋*; 大伴 真名歩; 圓谷 志郎; 松本 吉弘; Sorokin, P. B.*
Physical Review B, 86(8), p.085435_1 - 085435_7, 2012/08
被引用回数:55 パーセンタイル:86.45(Materials Science, Multidisciplinary)The graphane with chemically bonded alkali metals (Li, Na, K) was considered as potential material for hydrogen storage. The ab initio calculations show that such material can adsorb as many as four hydrogen molecules per Li, Na, and K metal atom. These values correspond to 12.20, 10.33, and 8.56 wt% of hydrogen, respectively, and exceed the DOE requirements. The thermodynamic analysis shows that Li-graphane complex is the most promising for hydrogen storage with ability to adsorb three hydrogen molecules per metal atom at 300 K and pressure in the range of 5-250 atm.
-Co films with current-perpendicular-to-plane geometry境 誠司; 三谷 誠司*; 松本 吉弘; 圓谷 志郎; Avramov, P.; 大伴 真名歩; 楢本 洋*; 高梨 弘毅
Journal of Magnetism and Magnetic Materials, 324(12), p.1970 - 1974, 2012/06
被引用回数:3 パーセンタイル:13.88(Materials Science, Multidisciplinary)Voltage-dependence of the tunneling magnetoresistance effect in the granular C-Co films has been investigated under the current-perpendicular-to-plane geometry. The transport measurements demonstrate that the granular C-Co films show unusually an exponential MR-V dependence. Small characteristic energies of less than 10's meV are derived from the temperature dependences of the characteristic voltage in the exponential relationship. Considering the voltage drops between Co nanoparticles and also the effect of cotunneling on the energy values, the characteristic energies for the voltage-induced degradation of the spin polarization are found to show a satisfactory agreement with that for the thermally-induced one. It can reasonably be expected that the onset of magnetic disorder to the localized d-electron spins at the interface region of the C-Co compound with Co nanoparticles will lead to the unusual voltage and temperature dependence of the MR ratio and the spin polarization.
for lithium batteries proposed by  simulations
simulationsKuzubov, A. A.*; Fedorov, A. S.*; Eliseeva, N. S.*; Tomilin, F. N.*; Avramov, P.; Fedorov, D. G.*
Physical Review B, 85(19), p.195415_1 - 195415_4, 2012/05
被引用回数:43 パーセンタイル:82.02(Materials Science, Multidisciplinary)The absorption energy and diffusion rates of lithium atoms inside graphitelike boron carbide (BC) crystal are investigated by the pseudopotential density-functional method using generalized gradient approximation. It is shown that lithium may effectively intercalate this structure with the maximum lithium concentration corresponding to LiBC stoichiometry, which is threefold in comparison to lithium in graphite. The potential barrier values for lithium diffusion both at low and maximum concentration are about 0.19 eV, so lithium atoms inside the BC structure can move easily. These findings suggest that boron carbide looks like a good candidate as an anode material in lithium ion batteries.
圓谷 志郎; 松本 吉弘; 大伴 真名歩; Avramov, P.; 楢本 洋*; 境 誠司
Journal of Applied Physics, 111(6), p.064324_1 - 064324_6, 2012/03
被引用回数:21 パーセンタイル:62.48(Physics, Applied)近年、グラフェンなどのナノカーボンを用いたスピントロニクスが注目されている。これらの材料ではスピン-軌道相互作用が小さいことからスピン拡散長が増大し、その結果高いスピン輸送特性の実現が期待されている。本論文では、グラフェンの成長条件の最適化を行い、グラフェン層数の精密制御法を確立した。サファイア基板上にNi(111)薄膜をエピタキシャル成長した。同試料を600 Cに保持した状態で前駆体となるベンゼンガスを曝露しグラフェンを成長した。グラフェンの層数・結晶構造は原子間力顕微鏡及びラマン分光によりそれぞれ評価した。単層・2層の精密制御を行うことにより、グラフェンへのドーピング状態が均一になることを明らかにした。さらに、電子エネルギー損失分光によりグラフェン/Ni(111)を調べた結果、同界面における強い相互作用の存在を明らかにした。
Cに保持した状態で前駆体となるベンゼンガスを曝露しグラフェンを成長した。グラフェンの層数・結晶構造は原子間力顕微鏡及びラマン分光によりそれぞれ評価した。単層・2層の精密制御を行うことにより、グラフェンへのドーピング状態が均一になることを明らかにした。さらに、電子エネルギー損失分光によりグラフェン/Ni(111)を調べた結果、同界面における強い相互作用の存在を明らかにした。
Pomogaev, V.*; Pomogaeva, A.*; Avramov, P.; Jalkanen, K. J.*; Kachin, S.*
Theoretical Chemistry Accounts, 130(4-6), p.609 - 632, 2011/12
被引用回数:10 パーセンタイル:22.74(Chemistry, Physical)Three polycyclic organic molecules in various solvents under thermo-dynamical influence were theoretically investigated using the recently developed statistical quantum mechanical/classical molecular dynamics (SQMMD) method for simulating electronic-vibrational spectra. The absorption bands of estradiol, benzene, and cyanoanthracene have been simulated, and most notably, the increase in the spectral intensity for the lowest excited state transition as the temperature is increased observed experimentally is well reproduced. In addition, this method has been extended to also treat luminescent processes, and it is seen that the experimental emission spectrum of cyanoanthracene is also well described. The method still needs further refinement, but results to date, including those presented in this work, document clearly that our model is one which is able to treat the many complex effects that the environment have on electronic absorption and emission spectra.
Fedorov, A. S.*; Fedorov, D. A.*; Kuzubov, A. A.*; Avramov, P.; 西村 好史*; Irle, S.*; Witek, H. A.*
Physical Review Letters, 107(17), p.175506_1 - 175506_5, 2011/10
被引用回数:23 パーセンタイル:73.01(Physics, Multidisciplinary)A methodology to evaluate the kinetic stability of carbon nanostructures is presented based on the assumption of the independent and random nature of thermal vibrations. The kinetic stability is directly correlated to the cleavage probability for the weakest bond of a given nanostructure. The application of the presented method to fullerenes and carbon nanotubes yields clear correlation to their experimentally observed relative isomer abundances. The general and simple formulation of the method ensures its applicability to other nanostructures for which formation is controlled by kinetic factors.
-Co compound/Ni bilayer structure松本 吉弘; 境 誠司; 圓谷 志郎; 高木 康多*; 中川 剛志*; 楢本 洋*; Avramov, P.; 横山 利彦*
Chemical Physics Letters, 511(1-3), p.68 - 72, 2011/07
被引用回数:5 パーセンタイル:15.54(Chemistry, Physical)C-Co化合物/Niの二層構造の電子・スピン状態についてX線磁気円二色性(XMCD)を用いた分光解析を行った。結果としてNi(111)表面上のC-Co化合物(数nm厚)では、Niの残留磁化のみで、局在スピン(Co dスピン)由来の強いXMCD信号が観測された。これはNi界面から3nm程度のC-Co化合物領域が、恐らく層間の電荷移動に関係のあるC分子を介した間接的交換相互作用により、下地のNiと強磁性的にカップリングしていることを指し示している。
 LC-DFT study of graphene, multilayer graphenes and graphite
LC-DFT study of graphene, multilayer graphenes and graphiteAvramov, P.; 境 誠司; 圓谷 志郎; 松本 吉弘; 楢本 洋*
Chemical Physics Letters, 508(1-3), p.86 - 89, 2011/05
被引用回数:25 パーセンタイル:64.10(Chemistry, Physical)Atomic structure of graphene, bi-, tri-, tetralayer graphenes and graphite as well was studied using HSE, LDA and PBE DFT approaches in periodic boundary conditions. Based on comparison of theoretical results with experimental data the performance of the methods was estimated. It was found that long-range corrected HSE potential is the most reliable DFT approximation to reproduce the atomic structure of weakly bound multilayer graphenes and graphite as well.
-Co films境 誠司; 三谷 誠司*; 菅井 勇; 高梨 弘毅; 松本 吉弘; 圓谷 志郎; 楢本 洋*; Avramov, P.; 前田 佳均
Physical Review B, 83(17), p.174422_1 - 174422_6, 2011/05
被引用回数:8 パーセンタイル:34.53(Materials Science, Multidisciplinary)Magnetotransport properties of the granular C-Co films are investigated over the broad bias voltage range from the region near zero bias by using the samples with the CPP (current-perpendicular-to-plane) geometry. It is revealed that the granular C-Co films show the I-V characteristics ascribed to cotunneling whose magnitudes are comparable to other molecular-based granular films. Furthermore, by considering the contribution of cotunneling on the magnitude of the TMR effect, it is successfully demonstrated that the tunnelling electrons generated at the interface between Co nanoparticles and a C-based matrix (C-Co compound) in the films have a significantly higher spin polarization (50-80%) than those in Co crystal and at the Al-oxide/Co interface. The present results clearly suggest that the high interfacial spin polarization is the most important cause of the large TMR effect in the granular C-Co films, in spite of the cotunneling-induced enhancement.
Chernozatonskii, L. A.*; Sorokin, P. B.*; Kuzubov, A. A.*; Sorokin, B. P.*; Kvashnin, A. G.*; Kvashnin, D. G.*; Avramov, P.; Yakobson, B. I.*
Journal of Physical Chemistry C, 115(1), p.132 - 136, 2011/01
被引用回数:88 パーセンタイル:89.20(Chemistry, Physical)The atomic structure and physical properties of quasi 2D diamanes (few-layered oriented diamond nanocrystals, covered by hydrogen atoms from both sides) are studied using electronic band structure calculations. It was shown that energy stability linear increases upon increasing of the thickness of proposed structures. All 2D carbon nanoclusters display direct dielectric band gaps with nonlinear quantum confinement response upon the thickness. Elastic properties of diamanes reveal complex dependence upon increasing of the number of 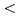 111
111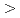 layers. All theoretical results were compared with available experimental data.
layers. All theoretical results were compared with available experimental data.
Avramov, P.; 巳波 壮馬*; Irle, S.*; Chernozatonskii, L. A.*; 諸熊 奎治*
Journal of Physical Chemistry C, 114(35), p.14692 - 14696, 2010/09
被引用回数:2 パーセンタイル:9.49(Chemistry, Physical)Atomic and electronic structure and energetic stability of newly proposed pentagonal and hexagonal chiral complex silicon nanowires (NWs) composed of five or six 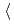 1 1 0
1 1 0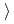 oriented crystalline fragments were studied using the ab initio DFT method. The chirality of the wires was caused by consecutive shifts of each fragment by 1/5 or 1/6 of the wire unit cell parameter and rotations of 4 and 3.3 for achiral pentagonal or hexagonal wires, respectively. Chirality causes the HOMO-LUMO gap to reduce by 0.1 eV. Chiral silicon nanowires are found to be metastable structures with a 4.5 (kcal/mol)/Si atom potential barrier for reversible chiral
oriented crystalline fragments were studied using the ab initio DFT method. The chirality of the wires was caused by consecutive shifts of each fragment by 1/5 or 1/6 of the wire unit cell parameter and rotations of 4 and 3.3 for achiral pentagonal or hexagonal wires, respectively. Chirality causes the HOMO-LUMO gap to reduce by 0.1 eV. Chiral silicon nanowires are found to be metastable structures with a 4.5 (kcal/mol)/Si atom potential barrier for reversible chiral 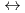 achiral transformation.
achiral transformation.
