


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
塚崎 克和*; 宮崎 直幸*; 松本 淳; 長江 成典*; 米村 重信*; 田ノ上 拓自*; 岩崎 憲治*; 竹市 雅俊*
Proceedings of the National Academy of Sciences of the United States of America, 111(45), p.16011 - 16016, 2014/11
被引用回数:38 パーセンタイル:63.74(Multidisciplinary Sciences)Fat and Dachsous cadherins regulate cell polarity and proliferation via their heterophilic interactions at intercellular junctions. Their ectodomains are unusually large because of the repetitive EC domains, which raises questions of how they fit in regular intercellular spaces. Cadherins typically exhibit a linear topology through the binding of Ca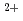 to the linker between the EC domains. Our electron microscopic observations of mammalian Fat4 and Dachsous1 ectodomains, however, revealed that, while their N-terminal regions exhibit a linear configuration, the C-terminal regions are kinked with multiple hairpin-like bends. Notably, certain EC-EC linkers in Fat4 and Dachsous1 lost Ca-binding amino acids. When such non-Ca-binding linkers were substituted for a normal linker in E-cadherin, the mutant E-cadherins deformed more extensively than the wild-type molecule. To simulate cadherin structures with non-Ca-binding linkers, we used the elastic network model and confirmed that bent configurations can be generated by deformation of the non-Ca-binding linkers. These findings suggest that Fat and Dachsous self-bend due to the loss of Ca-binding amino acids from specific EC-EC linkers, and therefore adapt to confined spaces.
to the linker between the EC domains. Our electron microscopic observations of mammalian Fat4 and Dachsous1 ectodomains, however, revealed that, while their N-terminal regions exhibit a linear configuration, the C-terminal regions are kinked with multiple hairpin-like bends. Notably, certain EC-EC linkers in Fat4 and Dachsous1 lost Ca-binding amino acids. When such non-Ca-binding linkers were substituted for a normal linker in E-cadherin, the mutant E-cadherins deformed more extensively than the wild-type molecule. To simulate cadherin structures with non-Ca-binding linkers, we used the elastic network model and confirmed that bent configurations can be generated by deformation of the non-Ca-binding linkers. These findings suggest that Fat and Dachsous self-bend due to the loss of Ca-binding amino acids from specific EC-EC linkers, and therefore adapt to confined spaces.
松本 淳; 鎌田 徹治*; 高木 淳一*; 岩崎 憲治*; 由良 敬
Biophysical Journal, 95(6), p.2895 - 2908, 2008/09
被引用回数:19 パーセンタイル:44.72(Biophysics)インテグリンは、多細胞生物において、細胞間の接着にかかわるタンパク質グループの総称である。インテグリンのなかには、活性化の際に、大きく構造を変化させるものがあることがわかっているが、その構造変化のメカニズムに関しては、よくわかっていなかった。われわれは、折りたたまれた構造をとっているインテグリンに対し、エラスティックネットワークモデルによる基準振動解析法を適用し、インテグリンの分子振動にとって重要な部位を発見した。さらなる計算の結果、その部位が、インテグリンの大規模な構造変化にとって重要であることを発見した。この重要性は、実験によっても確認した。さらに、さまざまな種類のインテグリンを調査し、重要な部位を構成するアミノ酸がどの程度保存されているかを調べたところ、限られたグループのインテグリンにおいてのみ、よく保存されていることを発見した。これは、大規模構造変化のメカニズムが、インテグリンの種類によって違うことを示している。
根本 慎一; 坂井 敏幸*; 算用子 裕孝; 菊池 憲治; 岩崎 伊佐央*; 栗林 正和*; 松島 和美*
PNC TN8410 93-283, 86 Pages, 1993/11
CPFにおけるホット試験は1982年9月30日に実施した高速炉使用済燃料ピンのせん断作業を皮切りに、これまでの約10年間、ピューレックス法を基本として高速炉燃料再処理に関した各プロセス試験を進めてきた。今回、これらのホット試験のうち、燃料の溶解試験に着目して総合的に評価・解析を加え、シミュレーションコードに反映できる溶解反応速度式を導出することができた。以下にその基本データについての概要を示す。高速炉使用済燃料の溶解速度は、反応表面積および系の硝酸濃度に比例する。また、温度に関してはアレニウスの式で補正できる。溶解速度=速度定数・反応表面積・(硝酸濃度)SUP1.7・e/SUP-E/RT (1)溶解速度は硝酸濃度の1.7乗に比例し、未照射UO ペレットの傾向とほぼ同じである。(2)アレニウスプロットにより求めた見かけの活性化エネルギーは11kcal/molであり、UOの溶解で報告されている同エネルギーにほぼ近い。(3)燃焼度の影響については、溶解反応速度式に反映できるような形での整理はできなかったが、溶解速度は硝酸濃度の低い系では燃焼度の増加に伴って低下する傾向にあること、また、8M程度の高濃度硝酸系では見かけ上ほぼ一定になることが観察された。(4)溶解速度の変化より溶解反応にかかわる有効表面積を推定し、せん断片および粉末の表面積変化を数式化した。(5)せん断片の"つぶれ"の影響については、約30%以上確保することによりほぼ一定の溶解速度を得ることができる。
ペレットの傾向とほぼ同じである。(2)アレニウスプロットにより求めた見かけの活性化エネルギーは11kcal/molであり、UOの溶解で報告されている同エネルギーにほぼ近い。(3)燃焼度の影響については、溶解反応速度式に反映できるような形での整理はできなかったが、溶解速度は硝酸濃度の低い系では燃焼度の増加に伴って低下する傾向にあること、また、8M程度の高濃度硝酸系では見かけ上ほぼ一定になることが観察された。(4)溶解速度の変化より溶解反応にかかわる有効表面積を推定し、せん断片および粉末の表面積変化を数式化した。(5)せん断片の"つぶれ"の影響については、約30%以上確保することによりほぼ一定の溶解速度を得ることができる。
松本 淳; 鎌田 徹治*; 岩崎 憲治*; 高木 淳一*; 由良 敬
no journal, ,
巨大な細胞外部分を持つ膜タンパク質であるインテグリンは、多細胞生物の細胞間接着に関与している。多くの種類のあるインテグリンのうち、活性化の際、大規模な構造変化を起こすものがあるが、その構造変化のメカニズムについては、ほとんどわかっていない。インテグリンのエラスティックネットワークモデルに対して、基準振動解析を行った結果、分子の内部運動に大きな影響のある相互作用を発見した。その相互作用に関与する部位の重要性は、実験でも確認された。
由良 敬; 鳴海 一成; 岩崎 憲治*
no journal, ,
放射線抵抗性細菌のDNA修復促進タンパク質PprAがどのようにしてDNA2本鎖切断修復を行っているかを明らかにすることを目的に、計算生物学の手法と電子顕微鏡単粒子観測を導入し、PprAタンパク質の立体構造に基づく修復機構の解明研究を行った。その結果、PprAタンパク質が4量体を形成した低分解能像を得ることに成功した。この像から、PprA単量体の構造は両端のおもりの大きさが異なる亜鈴を形成していると推定でき、4量体構造は亜鈴の柄部分が4個並行に樽状に並んでいると推定できた。次に、この構造を手がかりにして計算生物学の手法を駆使し、PprAタンパク質の原子分解能構造の推定を行った。その結果、PprAタンパク質のアミノ酸配列は、データベースにある立体構造中、RecJタンパク質の立体構造に一番よく適合することがわかった。また、PprAタンパク質の立体構造のどの部分でDNAと相互作用するかを推定したところ、亜鈴構造を特徴付ける中央のへリックスにDNAが相互作用することがわかってきた。
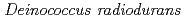 PprA protein
PprA protein鳴海 一成; 佐藤 勝也; 大庭 寛史; 由良 敬*; 岩崎 憲治*
no journal, ,
放射線抵抗性細菌から見いだした新規DNA修復促進タンパク質PprAのアミノ酸配列は、今まで知られているどのタンパク質のアミノ酸配列とも似ていない。また、PprAは、今まで知られているどのDNA結合モチーフも持ち合わせていない。そこで本研究では、PprAタンパク質のDNA結合様式に関する知見を得るために、電子顕微鏡単粒子解析を用いてPprAタンパク質の3次元構造を調べた。その結果、PprAタンパク質は、中心孔を有するダンベル型のホモ4量体を形成していることがわかった。この中心孔は、二本鎖DNA末端を包み込むのに十分な大きさであった。また、生物情報学的手法を用いてPprAタンパク質の3次元構造の推定を試みた結果、PprAタンパク質はダンベル型構造を持つ大腸菌RecJタンパク質の3次元構造に適合することが明らかになった。
松本 淳; 岩崎 憲治*; 高木 淳一*
no journal, ,
We have developed a computational approach to build an atomic model from an electron microscope (EM) image of proteins. In this approach, many atomic models are built first by deforming the X-ray crystal structure of the protein using a computational technique. Then, projection images of each atomic model to many different directions are generated. Finally, they are compared to the EM image. The atomic models with the projection images similar to the EM image are regarded as the candidates for the atomic structure of the protein from which the EM image was obtained. This approach was applied to the EM images of integrin. The X-ray crystal structure of integrin was solved for the closed conformation, while many EM images were obtained for the open conformation. The difference of the two conformations is so large that it is difficult to build the atomic model for the EM conformations by deforming the X-ray crystal structure using conventional computational techniques. Thus, we have used the coarse-grained model of the X-ray crystal structure to simulate the large-scale conformational change.
岩崎 憲治*; 松本 淳; 高木 淳一*
no journal, ,
We developed a method for comparing 2-D electron microscopy (EM) images to a pool of deformed atomic structures that reduces the susceptibility of subjective interpretation and is more expeditious than 3-D analysis. Firstly, we used normal mode analysis (NMA) in the context of an elastic network model (ENM) to prepare a pool of deformed structures from coarse-grain (CG) structural models obtained using Ca atom coordinates from crystal structures. Then, small deformations were applied to the model along low frequency normal modes. Both NMA and deformation were performed iteratively to build many different deformed atomic structures. 2-D images were then compared to these structures to objectively locate their model of best fit. The fitting-search is performed as a rigid body fitting, which works effectively for very noisy raw images as well as for molecules with a large conformational change. Our most representative example of this hybrid approach of using 2-D EM images and crystal structures has been the study of the integrin cell-adhesion molecules. Our hybrid method has allowed the investigation of the conformationally flexible integrins, and is providing useful evidence of whether other members of that family also use the switch-blade model observed in integrin avb3.
松本 淳; 高木 淳一*; 岩崎 憲治*
no journal, ,
We have developed a new approach to build a 3D atomic model from a single electron microscope (EM) image of a biomolecule. In this approach, the initial structure (X-ray crystal structure or modeled structure) of the molecule is deformed by computational techniques and many atomic models with different conformations are generated. The deformation is performed in such a way that the internal energy of the molecule does not increase so much. For this purpose, the lowest frequency normal modes are used in the deformation. Then, each atomic model is projected to many different directions, and each projection is fitted to the EM image and the fitting-score is calculated. The atomic models with high fitting-scores are the candidates that reproduce the EM image.
岩崎 憲治*; 高木 淳一*; 松本 淳
no journal, ,
受容体や細胞接着分子等、大きな細胞外領域を保有する膜タンパク質の構造は非常に柔軟で、中にはその機能との重要な相関を示唆する、大きなコンフォメーション変化を示すものも多い。こうした分子の構造は、電顕イメージング法では三次元構造を求めるのが非常に困難であり、得られたとしても低分解能で、生化学実験等に比して情報の乏しさは否めないものであった。そこで、二次元であれば、簡便にかつ迅速にその構造を吟味できることを提唱してきたが、その場合見た目に依存した主観的な議論にとどまることが常であった。今回、我々は、より客観的に解釈し、特定のアミノ酸配列についての吟味を可能にする方法を開発したのでここに報告する。電顕イメージに対して、そこに適切に当てはまるようにX線結晶構造解析で得られた原子座標を変化させるためには三次元であれ、二次元であれ解析イメージ自身に高い分解能が要求される。そこで松本らがこれまでに三次元用に開発した、あらかじめ様々に変化させた構造を発生させ、このうち最もよく当てはまるものを選出する方法を応用した。モデルタンパク質としてその詳細な構造と機能の相関が調べられているインテグリンavb3を用いて同方法を行ったところ、これまでの報告と一致する結果が得られた。そこで、異なるタンパク質やインテグリンにも適用し、生化学実験等で得られている結果と併せてその多様な構造が示す機能との相関を探った。
松本 淳; 高木 淳一*; 岩崎 憲治*
no journal, ,
We developed a method for comparing 2-D images to a pool of deformed atomic structures that reduces the susceptibility of subjective interpretation and is more expeditious than 3-D analysis. Firstly, we used normal mode analysis (NMA) in the context of an elastic network model to prepare a pool of deformed structures from coarse-grain (CG) structural models. Then, small deformations were applied to the model along low frequency normal modes. Both NMA and deformation were performed iteratively to build many different deformed atomic structures. 2-D images were then compared to these structures to objectively locate their model of best fit. The fitting-search is performed as a rigid body fitting, which works effectively for very noisy raw images as well as for molecules with a large conformational change. Our most representative example of this hybrid approach of using 2-D EM images and crystal structures has been the study of the integrin cell-adhesion molecules. Our hybrid method has allowed the investigation of the conformationally flexible integrins.
松本 淳; 高木 淳一*; 岩崎 憲治*
no journal, ,
We have developed a computational technique to build an atomic model from an electron microscopy (EM) image of a biological molecule. In this technique, many atomic models with different conformations of the molecule are prepared first by deforming the X-ray crystal structure or the modeled structure using a computational technique. Then, each atomic model is projected to many different directions and projection images are obtained. Finally, they are compared to the EM image. The atomic models with the projection images similar to the EM image are regarded as the candidates for the atomic structure of the molecule. This technique was applied to the EM images of integrin. Integrin is a membrane protein with a huge extracellular domain, and participates in cell-cell and cell-extracellular-matrix interactions. The X-ray crystal structure of the molecule is already solved. However, it is often very different form those seen in the EM images. By the conventional computational approach, it is difficult to reach the EM structure from the X-ray structure. Thus, we employed a coarse-grained model to simulate the large-scale conformational change.
松本 淳; 高木 淳一*; 岩崎 憲治*
no journal, ,
We have been developing a new computational approach to build a 3D atomic model from an electron microscope (EM) image of a biological molecule. In the usual approach, a 3D-EM model is constructed from many EM images. Then, a 3D atomic model is built mainly by deforming the X-ray crystal structure so that it fits into the 3D-EM model better. Our approach, on the other hand, uses only a single EM image (or an averaged image) and an X-ray crystal structure (or a modeled structure), and has the advantage of being applicable even to flexible molecules, for which it is difficult to construct 3D-EM models. We will report the results about the application to integrins, transmembrane proteins that involved in cell-cell and cell-extracellular matrix adhesions.
松本 淳; 高木 淳一*; 岩崎 憲治*
no journal, ,
We are developing a new computational approach to build a 3D atomic model from an electron microscope (EM) image of a biological molecule. In the usual approach, a 3D-EM model is constructed from many EM images. Then, a 3D atomic model is built mainly by deforming the X-ray crystal structure so that it fits into the 3D-EM model better. Our approach, on the other hand, uses only a single EM image (or an averaged image) and an X-ray crystal structure (or a modeled structure), and has the advantage of being applicable even to flexible molecules, for which it is difficult to construct 3D-EM models. We applied this approach to integrins, transmembrane proteins that involved in cell-cell and cell-extracellular matrix adhesions.
松本 淳; 高木 淳一*; 岩崎 憲治*
no journal, ,
生体分子の2次元電顕画像から原子分解能の3次元立体構造モデルを構築する計算手法の開発を行っている。通常のアプローチでは、多数の電顕画像から3次元電顕構造(3D-EM)を構築し、さらに、その構造に当てはまるようにX線結晶構造を変形することにより、原子分解能の3次元立体構造モデルを構築する。このアプローチでは、同じ立体構造を取っている多数の分子を様々な方向から撮影した電顕画像を用意する必要があるため、構造変化しやすい分子に対しては適用が難しい。そこで、我々は、射影像が電顕画像を再現するようにX線結晶構造を変形することにより、原子分解能の3次元立体構造モデルを構築する計算手法の開発を行っている。この計算手法では、1枚の電顕画像から1つの3次元立体構造を構築するため、構造変形しやすい分子に対しても適用が可能である。テスト計算の対象として、細胞接着タンパク質であるインテグリン V
V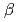 3を選んだ。インテグリンV3は、Caイオンを含む溶液中では折れ曲がった構造を取り、Mnイオンを含む溶液中では伸びた構造を取ることが、電子顕微鏡による観察でわかっている。一方、X線結晶構造は、Ca溶液中の構造に似ている。そのため、Mn溶液中の構造を再現するには、X線結晶構造の大規模な構造変形が必要である。構造変形計算を効率的に行うため、X線結晶構造を粗視化したモデルを用いた。
3を選んだ。インテグリンV3は、Caイオンを含む溶液中では折れ曲がった構造を取り、Mnイオンを含む溶液中では伸びた構造を取ることが、電子顕微鏡による観察でわかっている。一方、X線結晶構造は、Ca溶液中の構造に似ている。そのため、Mn溶液中の構造を再現するには、X線結晶構造の大規模な構造変形が必要である。構造変形計算を効率的に行うため、X線結晶構造を粗視化したモデルを用いた。

