


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
小林 恵太; 中村 博樹; 奥村 雅彦; 板倉 充洋; 町田 昌彦
Journal of Chemical Physics, 162(24), p.244508_1 - 244508_11, 2025/06
被引用回数:1 パーセンタイル:58.95(Chemistry, Physical)機械学習分子動力学法を用いて、(逆)蛍石構造における比熱異常の解析を行った。Farthest Point Sampling法とBootstrap法を活用し、効率的に第一原理教師データを生成することで、二酸化トリウム(蛍石構造)および酸化リチウム(逆蛍石構造)の機械学習ポテンシャルを構築した。これにより、機械学習分子動力学法は二酸化トリウムと酸化リチウムの報告されている熱物性を精度良く再現することが可能となった。これらの物質は、高温下において副格子の無秩序化に伴う比熱異常を示すが、そのメカニズムは複雑であり、完全には解明されていない。今回、液相・液相転移の解析に用いられる局所秩序変数の方法論を適用することで、(逆)蛍石構造における比熱異常が局所的な対称性の破れとして定式化できることを示した。
 -Fe
-Fe鈴土 知明; 海老原 健一; 都留 智仁; 森 英喜*
Journal of Applied Physics, 135(7), p.075102_1 - 075102_7, 2024/02
被引用回数:4 パーセンタイル:42.91(Physics, Applied)体心立方(bcc)金属および合金では延性脆性遷移温度以下において脆性的破壊が起きる。この事象は、脆性破壊を起こすき裂先端の臨界応力拡大係数が塑性変形を起こす臨界応力拡大係数よりも小さく塑性変形よりも脆性破壊が優先的に選択されるという考え方によって理論的に説明されている。この考え方は巨視的には正しいが、このような脆性破壊は常にき裂先端近傍での小規模な塑性変形、すなわちき裂先端塑性変形を伴う。この論文では、最近開発された-Feの機械学習原子間ポテンシャルを用いて原子論的モデリングを行い、この塑性の発現メカニズムを解析した。その結果、高速なき裂進展によってき裂先端位置の原子群が動的に活性化され、それがき裂先端塑性の前駆体になっていることが判明した。
藪内 聖皓*; 鈴土 知明
Journal of Nuclear Materials, 574, p.154161_1 - 154161_6, 2023/02
被引用回数:5 パーセンタイル:52.01(Materials Science, Multidisciplinary)原子炉材料において照射欠陥は機械的特性の劣化を引き起こす。これらの材料では、転位とボイドとの関係が機械的強度に特に重要である。これまで球状のボイドのみが研究されてきたが、本研究では球状ボイドと同時に観測されるファセット型ボイドに注目した。よって本研究では、純鉄の照射硬化におけるファセット型ボイドの効果を明らかにするために、分子動力学法を用いて解析した。具体的には、球状ボイドとファセット型ボイドの障害物強度と相互作用過程の違いや、ファセット型ボイドでも転位との結晶学的な配置関係によって相互作用に違いが出ることを明らかにした。
鈴土 知明; 海老原 健一; 都留 智仁; 森 英喜*
Scientific Reports (Internet), 12, p.19701_1 - 19701_10, 2022/11
被引用回数:13 パーセンタイル:55.92(Multidisciplinary Sciences)体心立方(bcc)遷移金属である-FeやWは、{110}面の表面エネルギーが最も低いにもかかわらず、{100}面に沿ってへき開割れが起きる。この奇妙な現象のメカニズムを解明するため、人工ニューラルネットワークの手法で作成した原子間ポテンシャルを用いて-Feの曲線のへき開き裂先端の大規模原子シミュレーションと直線のき裂先端の応力拡大係数解析を実施した。その結果、以下の新しい知見が得られた。{110}に沿ったへき開面のき裂先端から転位放出が観測され、そのことは{100}面が実際に起こるへき開面であることを示唆した。しかしながら、単純な直線状のき裂先端解析では、同じ結論は得られなかった。よって、機械的な強度を正しく予測するためには、高精度なポテンシャルを用いて、材料固有の複雑さを十分に捉えた原子論的なモデリングが必要であることが示唆された。本研究で採用した方法は、bcc遷移金属・合金のへき開問題に一般的に適用可能である。
小林 恵太; 山口 瑛子; 奥村 雅彦
Applied Clay Science, 228, p.106596_1 - 106596_11, 2022/10
被引用回数:15 パーセンタイル:78.69(Chemistry, Physical)粘土鉱物の一種であるカオリナイトの機械学習ポテンシャルを構築し、分子動力学法シミュレーションを実施することにより、カオリナイトの各種物性値を評価し、実験結果と比較した。その結果、原子構造や機械的特性を非常に良い精度で実験結果を再現することが確認された。さらに、構築した機械学習ポテンシャルを用いた分子動力学方シミュレーションで振動状態密度を評価したところ、古典分子動力学法や第一原理計算手法等の既存のシミュレーション手法では再現できなかった中性子非弾性散乱実験の結果をよく再現することがわかった。
 TEM observation and MD simulation of frank partial dislocation climbing in Al-Cu alloy
TEM observation and MD simulation of frank partial dislocation climbing in Al-Cu alloyChen, J.*; 吉田 健太*; 鈴土 知明; 嶋田 雄介*; 井上 耕治*; 今野 豊彦*; 永井 康介*
Materials Transactions, 63(4), p.468 - 474, 2022/04
被引用回数:3 パーセンタイル:17.54(Materials Science, Multidisciplinary)高分解能透過型電子顕微鏡(HRTEM)を用いてその場電子照射により、アルミニウム-銅(Al-Cu)合金のフランクループの発達を0.12nmの原子スケールの空間分解能で視覚化した。FCC-Al格子の[110]方向に沿ったその場HRTEM観察では、固有の積層欠陥の境界となるフランク部分転位は、純Al参照サンプルの場合とは異なり、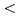 112
112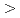 方向に沿って非対称の上昇を示した。この時の弾き出し損傷率は0.055-0.120dpa/sであった。分子動力学シミュレーションによって、この部分転位の非対称上昇はギニア-プレストンゾーン(GPゾーン)でのCu-Cu結合によるピン止め効果として説明された。
方向に沿って非対称の上昇を示した。この時の弾き出し損傷率は0.055-0.120dpa/sであった。分子動力学シミュレーションによって、この部分転位の非対称上昇はギニア-プレストンゾーン(GPゾーン)でのCu-Cu結合によるピン止め効果として説明された。
-iron; A Molecular dynamics study鈴土 知明; 海老原 健一; 都留 智仁
AIP Advances (Internet), 10(11), p.115209_1 - 115209_8, 2020/11
被引用回数:21 パーセンタイル:68.26(Nanoscience & Nanotechnology)BCC金属の脆性破壊のメカニズムはまだ明確には理解されているとは言えない。本研究では、鉄のへき開破壊の解析のため一連の3次元分子動力学シミュレーションを行った。特に、湾曲したき裂フロントから始まるモードI変形に焦点を当てた。シミュレーション結果、{100}面でへき開による脆性破壊が観察されたが、他の面では転位の放出によりき裂が鈍化した。この結果は{100}がbcc遷移金属で優先的に観測されるへ開面あるという一般的な実験的観察を再現した。
鈴土 知明; 高見澤 悠; 西山 裕孝; Caro, A.*; 外山 健*; 永井 康介*
Journal of Nuclear Materials, 540, p.152306_1 - 152306_10, 2020/11
被引用回数:13 パーセンタイル:73.11(Materials Science, Multidisciplinary)熱時効したFe-Cr合金はスピノーダル分解によって硬化を引き起こし、これはいわゆる475C脆性の直接的な原因である。スピノーダル分解が原子的相互作用によってどのように硬化を引き起こすのかを示すため、数値シミュレーションと実験を実施した。数値的な結果では、硬さが短距離秩序(SRO)パラメーターと比例することを示され、実験でもこの関係を統計誤差内で再現した。どちらの結果も、隣接するCr-Cr原子ペアが本質的に硬化を引き起こすことを示唆した。なぜなら、SROがそのようなペアの出現確率に一意的に依存しているからである。硬化の主な原因がそのようなCr-Crペア付近を通過する転位のピン止め効果であることが示唆されたが、このアイデアはさらなるモデリング研究により裏付けられた。
 5 grain boundary of -iron
5 grain boundary of -iron海老原 健一; 鈴土 知明
Proceedings of Joint International Conference on Supercomputing in Nuclear Applications + Monte Carlo 2020 (SNA + MC 2020), p.65 - 69, 2020/10
リンは鉄鋼材料において粒界脆化を引き起こす元素として知られている。さらに、照射による空孔や格子間原子の増加によってリン原子の粒界偏析が促進される。このことから、照射量や温度に対する粒界リン偏析量を評価するため、原子レベルの素過程に基づく拡散レート理論モデルを開発している。しかし、このモデルでは、粒界でのリンのトラップ及びデトラップモデルが適切に取り込まれていないため、実験結果と直接比較できる量を計算できない。粒界からのデトラップを考察するため、これまで、これまで鉄中の3対称傾角粒界内におけるリンの移動を分子動力学シミュレーションにより考察してきたが、本研究では、5でのリン移動をシミュレーションし、3との結果と比較した。その結果、800Kにおいて、3ではリン原子は比較的容易に鉄原子の間を移動するが、5では空孔がないと移動できないことが分かった。また、リン原子を置かない粒界領域中の鉄原子についても同様の傾向が見られた。これは、デトラップ過程をモデル化するための1つの知見を与えると考えられる。
WB-STEM observation of dislocation loop behavior in reactor pressure vessel steel during post-irradiation annealingDu, Y.*; 吉田 健太*; 嶋田 雄介*; 外山 健*; 井上 耕治*; 荒河 一渡*; 鈴土 知明; Milan, K. J.*; Gerard, R.*; 大貫 惣明*; et al.
Materialia, 12, p.100778_1 - 100778_10, 2020/08
長期に原子炉圧力容器の健全性を確保するためには、照射が材料に及ぼす影響を理解する必要がある。本研究では我々が新規開発したWB-STEMを用いて、中性子照射された原子炉圧力容器試験片を焼鈍中、照射誘起転位ループの観察を行った。焼鈍温度を上げると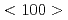 ループの割合が増加していることが確認された。また、2つの
ループの割合が増加していることが確認された。また、2つの
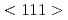 ループが衝突してループになる現象の観察に初めて成功した。転位に転位ループがデコレートする現象も観察され、分子動力学シミュレーションによってそのメカニズムが説明することができた。
ループが衝突してループになる現象の観察に初めて成功した。転位に転位ループがデコレートする現象も観察され、分子動力学シミュレーションによってそのメカニズムが説明することができた。
-iron海老原 健一; 鈴土 知明
TMS 2020; 149th Annual Meeting & Exhibition Supplemental Proceedings, p.995 - 1002, 2020/02
被引用回数:1 パーセンタイル:49.84(Materials Science, Multidisciplinary)リンは鉄鋼材料において粒界脆化を引き起こす元素として知られている。さらに、照射による空孔や格子間原子の増加によってリン原子の粒界偏析が促進される。このことから、照射量や温度に対する粒界リン偏析量を評価するため、原子レベルの素過程に基づく拡散レート理論モデルを開発している。しかし、このモデルでは、粒界でのリンのトラップ及びデトラップモデルが適切に取り込まれていないため、実験結果と直接比較できる量を計算できない。本研究では、粒界からのデトラップを考察するため、鉄中の3対称傾角粒界内におけるリンの移動を分子動力学でシミュレーションした。また、移動するリン原子を追跡し、その拡散障壁エネルギーを評価した。その結果、拡散障壁エネルギーは粒界の鉄原子の隙間の偏析サイトの偏析エネルギーと同程度であり、粒界中の鉄原子の間を移動することが分かった。これは、デトラップ過程をモデル化するための1つの知見を与えると考えられる。
3(111) symmetrical tilt grain boundary in -iron海老原 健一; 鈴土 知明
Modelling and Simulation in Materials Science and Engineering, 26(6), p.065005_1 - 065005_10, 2018/09
被引用回数:5 パーセンタイル:20.16(Materials Science, Multidisciplinary)照射誘起粒界リン偏析の見積もりは原子炉圧力容器鋼の脆化を評価する上で重要な要素であるが、粒界へのリン移動の物理的過程は依然として不明である。このことから、分子動力学を用いて3(111)対称傾角粒界へのリン移動を分子動力学シミュレーションによって評価した。結果として、粒界近傍 1nmの範囲で、自己格子間原子が粒界に押し出されることで、鉄-リン混合ダンベルのリン原子と八面体格子間リン原子が置換型原子になることを見出した。空孔-リン複合体も解離し、空孔はリン原子を引きずることなく粒界に吸収された。この結果は、従来考えられている偏析プロセスとは異なることから、それについて新しい視点が必要であると示唆している。
1nmの範囲で、自己格子間原子が粒界に押し出されることで、鉄-リン混合ダンベルのリン原子と八面体格子間リン原子が置換型原子になることを見出した。空孔-リン複合体も解離し、空孔はリン原子を引きずることなく粒界に吸収された。この結果は、従来考えられている偏析プロセスとは異なることから、それについて新しい視点が必要であると示唆している。
Lammers, L.*; Bourg, I. C.*; 奥村 雅彦; Kolluri, K.*; Sposito, G.*; 町田 昌彦
Journal of Colloid and Interface Science, 490, p.608 - 620, 2017/03
被引用回数:142 パーセンタイル:94.13(Chemistry, Physical)分子動力学法を用いる粘土鉱物シミュレーションは、これまで、無限系がほとんどであったが、本研究では、初めて粘土鉱物の微粒子の模型を作成し、セシウム吸着のシミュレーションを行った。その結果、微粒子のエッジ表面において、基盤表面では見られない強い吸着が起こることがわかった。特に、セシウムイオンとナトリウムイオンの強豪吸着では、エッジサイトはセシウムを選択的に吸着することがわかった。また、本研究で開発したエッジサイトの力場の構成法についても解説した。
冠城 雅晃; 山田 寛尚*; 宮川 毅*; 森河 良太*; 高須 昌子*; 加藤 宝光*; 上坂 充*
Polymer Journal, 48(2), p.189 - 195, 2016/02
被引用回数:5 パーセンタイル:16.25(Polymer Science)本研究は、テロメア一本鎖DNAとPOT1について分子動力学シミュレーションを100ns行った。テロメアDNAとPOT1の結合状態を確認するため、POT1の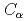 原子とテロメアDNAのO5'原子の距離を計算した。そして、単独状態と結合状態において、テロメア一本鎖DNAの両端塩基間距離、根平均二乗距離(RMSD)、慣性半径を計算した。さらに、単独状態と結合状態の根平均二乗揺らぎ(RMSF)を比較し、POT1とテロメアDNAの間の水素結合の平均数も計算した。グリニシン94(Gln94)と一本鎖テロメアDNAでPOT1と最近接なTTAGGGの一番目(G')のグアニンの間に水素結合が最頻度で現れる。そして、Gln94とG'が単独状態と結合状態でのRMSF値の差が最大になる。本研究では、Gln94とG'は、結合系において重要な部分で、結合状態の安定性に関係していると結論づけている。
原子とテロメアDNAのO5'原子の距離を計算した。そして、単独状態と結合状態において、テロメア一本鎖DNAの両端塩基間距離、根平均二乗距離(RMSD)、慣性半径を計算した。さらに、単独状態と結合状態の根平均二乗揺らぎ(RMSF)を比較し、POT1とテロメアDNAの間の水素結合の平均数も計算した。グリニシン94(Gln94)と一本鎖テロメアDNAでPOT1と最近接なTTAGGGの一番目(G')のグアニンの間に水素結合が最頻度で現れる。そして、Gln94とG'が単独状態と結合状態でのRMSF値の差が最大になる。本研究では、Gln94とG'は、結合系において重要な部分で、結合状態の安定性に関係していると結論づけている。
城地 保昌*; 北尾 彰朗*; 郷 信広
Journal of the American Chemical Society, 127(24), p.8705 - 8709, 2005/06
被引用回数:27 パーセンタイル:60.32(Chemistry, Multidisciplinary)ボゾンピークとは200K以下の生体高分子を含む多くのガラス状物質による非弾性中性子散乱やラマン散乱スペクトルの低振数領域に見られる幅の広いピークを指す。本論文では蛋白質のボゾンピークの起源に関する新しい描像を与える。分子動力学シミュレーションによれば、蛋白質分子のまわりの構造水分子は蛋白質のエネルギー地形の多極小性を一層際立たせ、これがボゾンピークの起源となっている。ピークは蛋白質分子が、水分子に由来するエネルギー極小構造に低温で束縛されることによって生じる。
清水 大志; 門吉 朋子; 蕪木 英雄; 山岸 信寛*; 長谷川 幸弘*; 樋口 健二
計算工学講演会論文集, 8(2), p.801 - 804, 2003/05
長時間計算が必要となるシミュレーションを分割する際の一連のリスタート処理について、ネットワーク上に分散した計算機群から利用可能な計算機を自動的に割り当てて実行する分散並列MDシミュレーション環境を構築した。約40万原子のシミュレーションでは、並列分子動力学ステンシルによるシミュレーションプログラムの可搬性とITBLの並列分散プログラム実行環境を組み合わせることにより、効率の良いシミュレーション計算の実行に非常に有効であることが確認された。
清水 大志; 蕪木 英雄
アンサンブル, (22), p.23 - 29, 2003/04
並列計算法開発グループでは、並列計算における基盤ソフトとして並列分子動力学シミュレーションツール「並列分子動力学ステンシル」(Parallel Molecular Dynamics Stencil)を開発した。「ステンシル」は材料物性研究等で広く使用される分子動力学法における並列プログラミングを効率的に進めるため、シミュレーションの対象となる系を記述するプログラムと並列化のためのプログラムを切り分けた後、再構成を行なったものである。本稿では「ステンシル」を用いたプログラミングの方法について具体例を挙げて説明する。
 O
O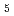 by MD simulation
by MD simulation鈴木 喜博*; 高瀬 桂一*; 秋山 功*; 鈴谷 賢太郎; 梅咲 則正*; 大鳥 範和*
Materials Transactions, 42(11), p.2242 - 2246, 2001/11
被引用回数:7 パーセンタイル:46.27(Materials Science, Multidisciplinary)POガラスの短範囲構造を、2体クーロン・ポテンシャルを用いた分子動力学シミュレーション法によって再現することに成功した。POガラスの最隣接原子相関P-Oは、P原子周りのO原子の配位数が4配位のPO 四面体を形成しており、そのうち3配位のO原子は他のPO四面体と頂点共有で架橋(bridging)している架橋酸素(Bridging Oxygen)O
四面体を形成しており、そのうち3配位のO原子は他のPO四面体と頂点共有で架橋(bridging)している架橋酸素(Bridging Oxygen)O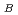 であり、残りの1配位のO原子は非架橋の酸素(terminal Oxygen)O
であり、残りの1配位のO原子は非架橋の酸素(terminal Oxygen)O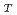 であることが構造化学的に予測され、パルス中性子回折実験の結果からもその短範囲構造は支持されている。われわれは、簡単な2体クーロン・ポテンシャルを用いた分子動力学シミュレーションを行い、パルス中性子回折実験による構造因子のhigh-Q側をよく再現する構造モデルを得た。このモデルの短範囲構造は、距離の長いP-Oが3つと距離の短いP-Oが1つからなる非対称なPO四面体構造を形成しており、その距離及び配位数は実験結果とよく一致した。このことは、POガラスの短範囲構造は、単に原子(イオン)の持つ電荷とサイズのみから決まっていることを意味している
であることが構造化学的に予測され、パルス中性子回折実験の結果からもその短範囲構造は支持されている。われわれは、簡単な2体クーロン・ポテンシャルを用いた分子動力学シミュレーションを行い、パルス中性子回折実験による構造因子のhigh-Q側をよく再現する構造モデルを得た。このモデルの短範囲構造は、距離の長いP-Oが3つと距離の短いP-Oが1つからなる非対称なPO四面体構造を形成しており、その距離及び配位数は実験結果とよく一致した。このことは、POガラスの短範囲構造は、単に原子(イオン)の持つ電荷とサイズのみから決まっていることを意味している
3(111)粒界および5(0-13)粒界中のリンの移動に関する考察海老原 健一; 鈴土 知明
no journal, ,
鉄鋼材料においてリン(P)原子は粒界に偏析し脆化を引き起こす。また照射環境下では粒界偏析が促進される。そのため、照射鉄鋼材料の高経年化において粒界P偏析量の評価は重要となる。粒界P偏析量を評価するため、これまで原子素過程に基づいて拡散レートモデルを開発してきた。モデルにおいて、Pの拡散に関する係数は原子レベルの計算に基づき比較的精度よく評価され組み込まれており、粒界におけるトラップ機構については原子レベルの考察を行っている。しかし、デトラップ機構については不明な点が多い。そのことから、今回、デトラップ機構の考察のため、BCC鉄の3(111)と5(0-13)の対称傾角粒界中におけるリンの移動を分子動力学でシミュレーションし、その違いを考察した。その結果、前者の粒界に比べ、後者ではリンが移動しにくいことが分かった。
鬼塚 貴志*; 大久保 学*; 鈴土 知明; 福元 謙一*
no journal, ,
原子炉の炉内構造物や圧力容器の中性子照射脆化の要因のひとつとして、ボイドなどの照射欠陥集合体が転位の運動を阻害して延性を低下させる可能性が指摘されている。しかしながら、ボイドがらせん転位に対してどの程度強い障害物になるのかということに関して、まだ十分に明らかになっていない。本研究では、鉄鋼材料の代替として純Feを研究対象に選択し、その中でのボイド形成が機械的性質に及ぼす影響を明らかにするため、分子動力学法を用いてボイド-らせん転位の相互作用の原子レベルのシミュレーションを行った。本報では特に、最大せん断応力の評価を行なったのでその結果について報告する。

