


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Ebihara, Kenichi; Fujihara, Hiro*; Shimizu, Kazuyuki*; Yamaguchi, Masatake; Toda, Hiroyuki*
International Journal of Hydrogen Energy, 136, p.751 - 756, 2025/06
Times Cited Count:1 Percentile:39.31(Chemistry, Physical)It has been experimentally reported that adding tin (Sn) to high-strength aluminum-zinc-magnesium (Al-Zn-Mg) alloys effectively suppresses hydrogen (H) embrittlement, which may be attributed to H absorption by the second-phase particles of Sn. To verify this fact, a simulation of H entry into the Sn phase in Al was performed using a model based on the reaction-diffusion equation that incorporates the solid solution energy of H evaluated by first-principles calculations. The results showed that the H solid solution site concentration of the second-phase particles must be at least five times higher than that of the Al phase for H absorption by the Sn second-phase particles to suppress H embrittlement. Therefore, the actual H embrittlement suppression effect of Sn second-phase particles is limited, and other factors may influence the suppression of H embrittlement in the experiment.
Noguchi, Yuji*; Aso, Seiyu*; Oyama, Kenji*; Ishigaki, Toru*; Yoneda, Yasuhiro; Matsuo, Hiroki*
Physical Review B, 111(21), p.214113_1 - 214113_13, 2025/06
Times Cited Count:2 Percentile:68.54(Materials Science, Multidisciplinary)We explore the electronic structure of high-quality bismuth sodium titanate (Bi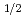 NaTiO
NaTiO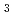 ) powders through a comprehensive approach combining the maximum entropy method (MEM)/Rietveld analysis of synchrotron radiation X-ray diffraction data collected at 200 K with density functional theory (DFT) calculations. We conclude that the covalent Bi-O bond and the resultant ferroelectricity stem primarily from the Bi-6p-O3-2p orbital interaction mediated through Ti-3d.
) powders through a comprehensive approach combining the maximum entropy method (MEM)/Rietveld analysis of synchrotron radiation X-ray diffraction data collected at 200 K with density functional theory (DFT) calculations. We conclude that the covalent Bi-O bond and the resultant ferroelectricity stem primarily from the Bi-6p-O3-2p orbital interaction mediated through Ti-3d.
Itakura, Mitsuhiro
AESJ-CSED Newsletter (Internet), (43), p.3 - 4, 2025/03
In recognition of receiving the Computational Science and Technology Division Achievement Award from the Atomic Energy Society, a summary of past research will be published as a preface.
Yamaguchi, Masatake; Ebihara, Kenichi; Tsuru, Tomohito; Itakura, Mitsuhiro
Materials Transactions, 64(11), p.2553 - 2559, 2023/11
Times Cited Count:11 Percentile:66.32(Materials Science, Multidisciplinary)We attempted to calculate the hydrogen trapping energies on the incoherent interfaces of MgZn precipitates and MgSi crystallites in aluminum alloys from first-principles calculations. Since the unit cell containing the incoherent interface does not satisfy the periodic boundary condition, resulting in a discontinuity of crystal blocks, the hydrogen trapping energy was calculated in a region far from the discontinuity (vacuum) region. We found considerable trapping energies for hydrogen atoms at the incoherent interfaces consisting of assumed atomistic arrangement. We also conducted preliminary calculations of the reduction in the cohesive energy by hydrogen trapping on the incoherent interfaces of MgSi in the aluminum matrix.
precipitates and MgSi crystallites in aluminum alloys from first-principles calculations. Since the unit cell containing the incoherent interface does not satisfy the periodic boundary condition, resulting in a discontinuity of crystal blocks, the hydrogen trapping energy was calculated in a region far from the discontinuity (vacuum) region. We found considerable trapping energies for hydrogen atoms at the incoherent interfaces consisting of assumed atomistic arrangement. We also conducted preliminary calculations of the reduction in the cohesive energy by hydrogen trapping on the incoherent interfaces of MgSi in the aluminum matrix.
Kobayashi, Keita; Okumura, Masahiko; Nakamura, Hiroki; Itakura, Mitsuhiro; Machida, Masahiko; Urata, Shingo*; Suzuya, Kentaro
Scientific Reports (Internet), 13, p.18721_1 - 18721_12, 2023/11
Times Cited Count:21 Percentile:80.65(Multidisciplinary Sciences)The first sharp peak diffraction peak (FSDP) in the structure factor of amorphous materials is thought to reflect the medium-range order structure in amorphous materials, and the structural origin of the FSDP has been a subject of ongoing debate. In this study, we employed machine learning molecular dynamics (MLMD) with nearly first-principles calculation accuracy to investigate the structural origin of the FSDP in high-density silica glass. First, we successfully reproduced various experimental data of high-density silica glass using MLMD. Furthermore, we revealed that the development (or reduction) of the FSDP in high-density silica glass is characterized by the deformation behavior of ring structures in Si-O covalent bond networks under compression.
Tatsumi, Kazuyoshi; Okudaira, Takuya*; Kofu, Maiko; Rozyczko, P.*
Journal of Physics; Condensed Matter, 36(37), p.375901_1 - 375901_13, 2023/06
Times Cited Count:0 Percentile:0.00(Physics, Condensed Matter)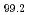 Zn
Zn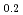 Y
Y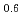 alloy using X-ray photoelectron spectroscopy
alloy using X-ray photoelectron spectroscopyMiyazaki, Hidetoshi*; Akatsuka, Tatsuyoshi*; Kimura, Koji*; Egusa, Daisuke*; Sato, Yohei*; Itakura, Mitsuhiro; Takagi, Yasumasa*; Yasui, Akira*; Ozawa, Kenichi*; Mase, Kazuhiko*; et al.
Materials Transactions, 64(6), p.1194 - 1198, 2023/06
Times Cited Count:1 Percentile:6.78(Materials Science, Multidisciplinary)We investigated the electronic structure of the MgZnY alloy using hard and soft X-ray photoemission spectroscopy and electronic band structure calculations to understand the mechanism of the phase stability of this material. Electronic structure of the MgZnY alloy showed a semi-metallic electronic structure with a pseudo-gap at the Fermi level. The observed electronic structure of the MgZnY alloy suggests that the presence of a pseudogap structure is responsible for phase stability.
Sato, Yohei*; Egusa, Daisuke*; Miyazaki, Hidetoshi*; Kimura, Koji*; Itakura, Mitsuhiro; Terauchi, Masami*; Abe, Eiji*
Materials Transactions, 64(5), p.950 - 954, 2023/05
Times Cited Count:2 Percentile:6.78(Materials Science, Multidisciplinary)Dilute Mg-Zn-Y alloy with a mille-feuille structure (MFS) exhibits a mechanical strength comparable to Mg-Zn-Y alloy with long period stacking/ordered (LPSO) structure through kink deformation. In order to deepen understanding the thermal stability of the MFS-type Mg alloys, it is required to clarify the solute cluster structures composed of Zn and Y in solute enriched stacking faults (SESFs). In this study, electron energy-loss and energy dispersive X-ray spectroscopy based on scanning transmission electron microscopy (STEM-EELS/EDS) were conducted to investigate the electronic structure and composition of Zn and Y in the SESFs of the MFS-Mg alloy. Zn-L2,3 spectra indicated that the valence charges of Zn in the dilute Mg alloy were different from that of the LPSO-type Mg-Zn-Y alloy. In addition, the intensity ratio of L3/L2 in Y-L2,3 spectrum of the dilute MFS-Mg alloy was larger than that of the LPSO-Mg alloy, reflecting the electron occupancies of 4d3/2 and 4d5/2 orbitals of Y atoms were different from those of the LPSO-Mg alloys. STEM-EELS analysis of the SESF composition in the dilute MFS-Mg alloy indicated that the Zn/Y ratio should be lower than that of the LPSO-Mg alloy, which was confirmed also by STEM-EDS measurements. These results indicate that the cluster structure in the SESFs of the dilute MFS-Mg alloy should be different from the ideal Zn6Y8 cluster in the LPSO-type Mg-Zn-Y alloys.
Itakura, Mitsuhiro; Yamaguchi, Masatake; Egusa, Daisuke*; Abe, Eiji*
Materials Transactions, 64(4), p.813 - 816, 2023/04
Times Cited Count:4 Percentile:30.19(Materials Science, Multidisciplinary)Kobayashi, Keita; Nakamura, Hiroki; Itakura, Mitsuhiro; Machida, Masahiko; Okumura, Masahiko
Materia, 62(3), p.175 - 181, 2023/03
no abstracts in English
Kobayashi, Keita; Okumura, Masahiko; Nakamura, Hiroki; Itakura, Mitsuhiro; Machida, Masahiko; Cooper, M. W. D.*
Scientific Reports (Internet), 12(1), p.9808_1 - 9808_11, 2022/06
Times Cited Count:24 Percentile:77.92(Multidisciplinary Sciences)no abstracts in English
Igarashi, Takahiro; Otani, Kyohei; Komatsu, Atsushi; Kato, Chiaki; Sakairi, Masatoshi*
Bosei Kanri, 66(4), p.141 - 145, 2022/04
Metal corrosion is a material deterioration phenomenon based on electrochemical reactions on an atomic scale. In this paper, various methods for acquiring physical properties on metal surfaces using first-principles calculations were described. As examples of applying first-principles calculation to metal corrosion, the effect of hydrogen adsorption on the metal surface on the potential change and the effect of cation atoms in the aqueous solution on the corrosion resistance of the metal were reported.
 -clustering in atomic nuclei from first principles with statistical learning and the Hoyle state character
-clustering in atomic nuclei from first principles with statistical learning and the Hoyle state characterOtsuka, Takaharu; Abe, Takashi*; Yoshida, Toru*; Tsunoda, Yusuke*; Shimizu, Noritaka*; Itagaki, Naoyuki*; Utsuno, Yutaka; Vary, J. P.*; Maris, P.*; Ueno, Hideki*
Nature Communications (Internet), 13, p.2234_1 - 2234_10, 2022/04
Times Cited Count:76 Percentile:98.16(Multidisciplinary Sciences)no abstracts in English
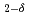 CoGa
CoGa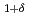 films
filmsKubota, Takahide*; Takano, Daichi*; Kota, Yohei*; Mohanty, S.*; Ito, Keita*; Matsuki, Mitsuhiro*; Hayashida, Masahiro*; Sun, M.*; Takeda, Yukiharu; Saito, Yuji; et al.
Physical Review Materials (Internet), 6(4), p.044405_1 - 044405_12, 2022/04
Times Cited Count:7 Percentile:35.80(Materials Science, Multidisciplinary)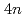 self-conjugate nuclei in
self-conjugate nuclei in  no-core Monte Carlo shell model calculations with nonlocal
no-core Monte Carlo shell model calculations with nonlocal 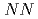 interactions
interactionsAbe, Takashi*; Maris, P.*; Otsuka, Takaharu*; Shimizu, Noritaka*; Utsuno, Yutaka; Vary, J. P.*
Physical Review C, 104(5), p.054315_1 - 054315_22, 2021/11
Times Cited Count:13 Percentile:76.80(Physics, Nuclear)no abstracts in English
Kobayashi, Keita; Nagai, Yuki; Itakura, Mitsuhiro; Shiga, Motoyuki
Journal of Chemical Physics, 155(3), p.034106_1 - 034106_9, 2021/07
Times Cited Count:15 Percentile:67.20(Chemistry, Physical)no abstracts in English
Suzudo, Tomoaki; Tsuru, Tomohito
AIP Advances (Internet), 11(6), p.065012_1 - 065012_7, 2021/06
Times Cited Count:6 Percentile:30.30(Nanoscience & Nanotechnology)In the current study, we analyzed the self-interstitial atoms (SIAs) in BCC molybdenum (Mo) and tungsten (W) in comparison with other BCC transition metals utilizing first-principles method; particularly, we focused on uncommon dumbbells, whose direction are inclined from 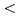 111
111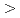 toward 110 on the {110} plane. Such a direction is not stable neither in the group 5 BCC metals (i.e., vanadium, niobium, and tantalum) nor in -iron. Our first-principles relaxation simulations indicated that inclined dumbbells were more energetically-favored than common 111 dumbbells in Mo, while this is not necessarily the case for W. However, under a certain degree of lattice strain, such as shear or expansive strain, could make inclined dumbbells more favored also in W, suggesting that the lattice strain can substantially influence the migration barrier of SIAs in these metals because inclined dumbbells generally have a larger migration barrier than 111 dumbbells.
toward 110 on the {110} plane. Such a direction is not stable neither in the group 5 BCC metals (i.e., vanadium, niobium, and tantalum) nor in -iron. Our first-principles relaxation simulations indicated that inclined dumbbells were more energetically-favored than common 111 dumbbells in Mo, while this is not necessarily the case for W. However, under a certain degree of lattice strain, such as shear or expansive strain, could make inclined dumbbells more favored also in W, suggesting that the lattice strain can substantially influence the migration barrier of SIAs in these metals because inclined dumbbells generally have a larger migration barrier than 111 dumbbells.
Kobayashi, Keita; Nakamura, Hiroki; Yamaguchi, Akiko; Itakura, Mitsuhiro; Machida, Masahiko; Okumura, Masahiko
Computational Materials Science, 188, p.110173_1 - 110173_14, 2021/02
Times Cited Count:33 Percentile:79.47(Materials Science, Multidisciplinary)no abstracts in English
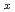 U
U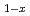 O
OItakura, Mitsuhiro; Nakamura, Hiroki; Kitagaki, Toru; Hoshino, Takanori; Machida, Masahiko
Journal of Nuclear Science and Technology, 56(9-10), p.915 - 921, 2019/09
Times Cited Count:2 Percentile:15.62(Nuclear Science & Technology)To elucidate the mechanical properties of fuel debris inside the Fukushima Daiichi Nuclear Power Plant, we use first-principles calculations to evaluate mechanical properties of cubic Zr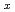 UO
UO , which is a main component of the fuel debris. We focus on the dependence of mechanical properties on the fraction x of zirconium, compare our results with recent experiment of simulated debris, in which dependences of elastic moduli and fracture toughness on the ZrO content showed deviation from a simple linear relation. We show that elastic moduli drop at around x=0.25 and increase again for larger values of x, as has been observed in experiments. The reason of the drop is a softening owing to disordered atomistic structures induced by the solute zirconium atoms. We also find that stress-strain curves for the x=0.125 case show marked hysteresis owing to the existence of many meta-stable states. We show that this hysteresis leads to slightly increased fracture toughness, but it is not enough to account for the significant increase of fracture toughness observed in experiments.
, which is a main component of the fuel debris. We focus on the dependence of mechanical properties on the fraction x of zirconium, compare our results with recent experiment of simulated debris, in which dependences of elastic moduli and fracture toughness on the ZrO content showed deviation from a simple linear relation. We show that elastic moduli drop at around x=0.25 and increase again for larger values of x, as has been observed in experiments. The reason of the drop is a softening owing to disordered atomistic structures induced by the solute zirconium atoms. We also find that stress-strain curves for the x=0.125 case show marked hysteresis owing to the existence of many meta-stable states. We show that this hysteresis leads to slightly increased fracture toughness, but it is not enough to account for the significant increase of fracture toughness observed in experiments.
 Cs solid-state NMR and density functional theory calculations
Cs solid-state NMR and density functional theory calculationsOkubo, Takahiro*; Okamoto, Takuya*; Kawamura, Katsuyuki*; Gu gan, R.*; Deguchi, Kenzo*; Oki, Shinobu*; Shimizu, Tadashi*; Tachi, Yukio; Iwadate, Yasuhiko*
gan, R.*; Deguchi, Kenzo*; Oki, Shinobu*; Shimizu, Tadashi*; Tachi, Yukio; Iwadate, Yasuhiko*
Journal of Physical Chemistry A, 122(48), p.9326 - 9337, 2018/12
Times Cited Count:19 Percentile:63.07(Chemistry, Physical)The structures of Cs adsorption on montmorillonite were investigated by the nuclear magnetic resonance (NMR) spectroscopy. The NMR spectra of Cs adsorbed on montmorillonite samples were measured under different Cs contents and relative humidity levels. NMR parameters were evaluated by the first principle calculations in order to identify the relationship between adsorbed Cs structures and NMR parameters. The comparisons between experimental and theoretical NMR spectra revealed that Cs is preferentially adsorbed at sites near Al for low Cs substituted montmorillonites, and that non-hydrated Cs present in partially Cs substituted samples, even after being hydrated under high relative humidity.

