


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Ohashi, Tomonori*; Sakamaki, Tatsuya*; Funakoshi, Kenichi*; Steinle-Neumann, G.*; Hattori, Takanori; Yuan, L.*; Suzuki, Akio*
Journal of Mineralogical and Petrological Sciences (Internet), 120(1), p.240926a_1 - 240926a_13, 2025/06
We explore the structures of dry and hydrated (H O and DO) Na
O and DO) Na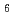 Si
Si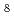 O
O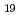 melt at 0-6 GPa and 1000-1300 K and glasses recovered from high pressure and temperatures by in-situ neutron and X-ray diffraction. The structures of the melts at 0-10 GPa and 3000 K are also investigated by ab-initio molecular dynamics simulation. In-situ neutron experiments revealed that the D-O distance increases with compression due to the formation of -O-D-O- bridging species, which is reproduced by the molecular dynamics simulations. The pressure-induced -O-D-O- formation reflects a more rigid incorporation of hydrogen, which acts as a mechanism for the experimentally observed higher solubility of water in silicate melts. Together with shrinking modifier domains, this process dominates the compression behavior of hydrous NaSiO melt, whereas the compression of dry NaSiO at 0-10 GPa and 3000 K is governed largely by bending of the Si-O-Si angle. The molecular dynamics simulations on hydrous NaSiO melts further suggest that the sodium ions are scavenged from its network-modifying role via 2(
melt at 0-6 GPa and 1000-1300 K and glasses recovered from high pressure and temperatures by in-situ neutron and X-ray diffraction. The structures of the melts at 0-10 GPa and 3000 K are also investigated by ab-initio molecular dynamics simulation. In-situ neutron experiments revealed that the D-O distance increases with compression due to the formation of -O-D-O- bridging species, which is reproduced by the molecular dynamics simulations. The pressure-induced -O-D-O- formation reflects a more rigid incorporation of hydrogen, which acts as a mechanism for the experimentally observed higher solubility of water in silicate melts. Together with shrinking modifier domains, this process dominates the compression behavior of hydrous NaSiO melt, whereas the compression of dry NaSiO at 0-10 GPa and 3000 K is governed largely by bending of the Si-O-Si angle. The molecular dynamics simulations on hydrous NaSiO melts further suggest that the sodium ions are scavenged from its network-modifying role via 2(![$$^{[4]}$$](/search/images/ERPHWWZULV4SI.gif) Si-O
Si-O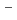 + Na
+ Na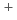 )
)  Si-(O-
Si-(O-![$$^{[5]}$$](/search/images/ERPHWWZVLV4SI.gif) Si-O)
Si-O)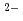 + 2Na and Si-O + Na + Si-OH Si-(O-H-O-Si) + Na with increasing pressure.
+ 2Na and Si-O + Na + Si-OH Si-(O-H-O-Si) + Na with increasing pressure.
Okita, Shoichiro; Goto, Minoru
Proceedings of 12th International Conference on Nuclear Criticality Safety (ICNC2023) (Internet), 10 Pages, 2023/10
Yamamoto, Tomohiko; Kato, Atsushi; Hayakawa, Masato; Shimoyama, Kazuhito; Ara, Kuniaki; Hatakeyama, Nozomu*; Yamauchi, Kanau*; Eda, Yuhei*; Yui, Masahiro*
Proceedings of 2023 International Congress on Advanced in Nuclear Power Plants (ICAPP 2023) (Internet), 6 Pages, 2023/04
Yotsuji, Kenji*; Tachi, Yukio; Sakuma, Hiroshi*; Kawamura, Katsuyuki*
Applied Clay Science, 204, p.106034_1 - 106034_13, 2021/04
Times Cited Count:102 Percentile:99.77(Chemistry, Physical)Gonzal z, M. A.*; Borodin, O.*; Kofu, Maiko; Shibata, Kaoru; Yamada, Takeshi*; Yamamuro, Osamu*; Xu, K.*; Price, D. L.*; Saboungi, M.-L.*
z, M. A.*; Borodin, O.*; Kofu, Maiko; Shibata, Kaoru; Yamada, Takeshi*; Yamamuro, Osamu*; Xu, K.*; Price, D. L.*; Saboungi, M.-L.*
Journal of Physical Chemistry Letters (Internet), 11(17), p.7279 - 7284, 2020/09
Times Cited Count:24 Percentile:80.73(Chemistry, Physical)Ichihara, Akira
JAEA-Review 2019-046, 36 Pages, 2020/03
 JAEA-Review-2019-046.pdf:1.55MB
JAEA-Review-2019-046.pdf:1.55MB
Toward the revision of JENDL-4.0, we conducted a literature survey on how to compute the cross section of thermal neutrons scattered by a liquid. This report summarizes the computational methods for evaluating thermal neutron cross sections with molecular dynamics simulations. The cross section can be expressed with a function called as scattering law. For light and heavy water, the scattering law data instead of the cross sections have been provided in nuclear databases. In this report we review the formulations of the scattering laws. The scattering laws can be derived from both the intermediate scattering function and the space-time correlation function. Features of the derived scattering laws are briefly explained. It is shown that the scattering law data can be evaluated using a molecular dynamics simulation of the liquid that is the target of thermal neutrons.
Yotsuji, Kenji*; Tachi, Yukio; Kawamura, Katsuyuki*; Arima, Tatsumi*; Sakuma, Hiroshi*
Nendo Kagaku, 58(1), p.8 - 25, 2019/00
Molecular dynamics (MD) simulations were conducted to investigate physical properties of water and cations in montmorillonite interlayer nanopores. The swelling behaviors and hydration states were firstly evaluated as functions of interlayer cations and layer charge. The diffusion coefficients of water and cations in interlayer nanopores were decreased in comparison with those in bulk water and came closer to those in bulk water when basal spacing increased. The viscosity coefficients of interlayer water estimated indicated a significant effect of viscoelectricity at 1- and 2-layer hydration states and higher layer charge of montmorillonite. These trends from MD calculations were confirmed to be consistent with existing measured data and previous MD simulation. In addition, model and parameter related to viscoelectric effect used in the diffusion model was refined based on comparative discussion between MD simulations and measurements. The series of MD calculations could provide atomic level understanding for the developments and improvements of the diffusion model for compacted montmorillonite.
Arima, Tatsumi*; Idemitsu, Kazuya*; Inagaki, Yaohiro*; Kawamura, Katsuyuki*; Tachi, Yukio; Yotsuji, Kenji
Progress in Nuclear Energy, 92, p.286 - 297, 2016/09
Times Cited Count:15 Percentile:77.12(Nuclear Science & Technology)Diffusion and adsorption behavior of uranyl (UO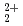 ) species is important for the performance assessment of radioactive waste disposal. The diffusion behaviors of UO, K
) species is important for the performance assessment of radioactive waste disposal. The diffusion behaviors of UO, K , CO
, CO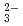 and Cl
and Cl and H
and H O in the aqueous solutions were evaluated by molecular dynamics (MD) calculations. The diffusion coefficient (De) of UO is the smallest and is 26% less than the self-diffusion coefficient of HO. For the aqueous solution with high concentration of carbonate ions, uranyl carbonate complexes: UOCO
O in the aqueous solutions were evaluated by molecular dynamics (MD) calculations. The diffusion coefficient (De) of UO is the smallest and is 26% less than the self-diffusion coefficient of HO. For the aqueous solution with high concentration of carbonate ions, uranyl carbonate complexes: UOCO and UO(CO) can be observed. For the clay (montmorillonite or illite)-aqueous solution systems, the adsorption and diffusion behaviors of UO and K were evaluated by MD calculations. The distribution coefficients (Kd) increase with the layer charge of clay, and Kd of UO might be smaller than that of K. Further, their two-dimensional diffusion coefficients were relatively small in the adsorption layer and were extremely small for illite with higher layer charge.
and UO(CO) can be observed. For the clay (montmorillonite or illite)-aqueous solution systems, the adsorption and diffusion behaviors of UO and K were evaluated by MD calculations. The distribution coefficients (Kd) increase with the layer charge of clay, and Kd of UO might be smaller than that of K. Further, their two-dimensional diffusion coefficients were relatively small in the adsorption layer and were extremely small for illite with higher layer charge.
Herv du Penhoat, M.-A.*; Kamol Ghose, K.*; Gaigeot, M.-P.*; Vuilleumier, R.*; Fujii, Kentaro; Yokoya, Akinari; Politis, M.-F.*
Physical Chemistry Chemical Physics, 17(48), p.32375 - 32383, 2015/12
Times Cited Count:9 Percentile:32.56(Chemistry, Physical)Ikeda, Takashi; Hou, Z.*; Chai, G.-L.*; Terakura, Kiyoyuki*
Hyomen Kagaku, 36(7), p.345 - 350, 2015/07
Carbon alloy catalysts (CACs) are one of promising candidates for platinum-substitute cathode catalysts for polymer electrolyte fuel cells. We have investigated possible mechanisms of oxygen reduction reactions (ORRs) for CACs via first-principles-based molecular dynamics simulations. In this contribution, we review possible ORRs at likely catalytic sites of CACs suggested from our simulations.
Ikeda, Takashi
Journal of Chemical Physics, 141(4), p.044501_1 - 044501_8, 2014/07
Times Cited Count:17 Percentile:53.39(Chemistry, Physical)From both the polarized and depolarized Raman scattering spectra of supercritical water a peak located at around 1600 cm , attributed normally to bending mode of water molecules, was experimentally observed to vanish, whereas the corresponding peak remains clearly visible in the measured infrared (IR) absorption spectrum. In this computational study a theoretical formulation for analyzing the IR and Raman spectra is developed via first principles molecular dynamics combined with the modern polarization theory. We demonstrate that the experimentally observed peculiar behavior of the IR and Raman spectra for water are well reproduced in our computational scheme. We discuss the origins of a feature observed at 1600 cm in Raman spectra of ambient water.
, attributed normally to bending mode of water molecules, was experimentally observed to vanish, whereas the corresponding peak remains clearly visible in the measured infrared (IR) absorption spectrum. In this computational study a theoretical formulation for analyzing the IR and Raman spectra is developed via first principles molecular dynamics combined with the modern polarization theory. We demonstrate that the experimentally observed peculiar behavior of the IR and Raman spectra for water are well reproduced in our computational scheme. We discuss the origins of a feature observed at 1600 cm in Raman spectra of ambient water.
Kotulic Bunta, J.*; Laaksonen, A.*; Pinak, M.; Nemoto, Toshiyuki*
Computational Biology and Chemistry, 30(2), p.112 - 119, 2006/04
Times Cited Count:5 Percentile:30.38(Biology)Due to their lethal consequences, single and double strand breaks are among the most important and dangerous DNA lesions. This work defines and analyzes a DNA with single strand break as a template study for future complex analyses of biologically serious double strand break damage and its enzymatic repair mechanisms. Besides a non-damaged DNA serving as a reference system, system with open valences of the atoms at the strand break ends as well as a system with filled valences was simulated. As for the results, the system with open valences is partly disrupted, and the system with filled valences is stabilized by forming new hydrogen bonds between two strand endings.
Pinak, M.
Modern Methods for Theoretical Physical Chemistry of Biopolymers, p.191 - 210, 2006/00
Results of molecular dynamics (MD) studies of several lesions on DNA and their respective repair enzymes are presented. Main focus is to describe structural and energy changes in DNA molecule with the respect to proper recognition of the lesion by respective repair enzyme. Pyrimidine and purine lesions were studied using MD simulation code Amber and respective force field modified for each lesion. The significant structural changes as breaking of hydrogen bonds network opening and bending the DNA double helix were observed. This collapsing of the double helical structure around the lesion is considered to facilitate docking of the repair enzyme into the DNA. Specific values of electrostatic interaction energy that enables repair enzyme to discriminate lesion from non-damaged site were also detected at several lesion sites.
 and La compared; An Application of metadynamics
and La compared; An Application of metadynamicsIkeda, Takashi; Hirata, Masaru; Kimura, Takaumi
Journal of Chemical Physics, 122(24), p.244507_1 - 244507_5, 2005/06
Times Cited Count:51 Percentile:83.51(Chemistry, Physical)We studied the hydration structures of Y and La in aqueous solutions by applying the metadynamics method recently introduced as a tool to explore reaction pathways based on the Car-Parrinello molecular dynamics. By employing the metal-water oxygen coordination number as a collective variable of the metadynamics a couple of aqua and chloro-aqua complexes are successfully generated within the time scales of 10 ps. The reconstructed free energy surface captures the characteristics of the hydration of the light and heavy trivalent rare-earth ions. The present study demonstrates that the metadynamics based on the Car-Parrinello molecular dynamics is a promising tool for exploring the free energy surface of complicated systems such as solutions.
Fujimoto, Hirofumi*; Pinak, M.; Nemoto, Toshiyuki*; O'Neill, P.*; Kume, Etsuo; Saito, Kimiaki; Maekawa, Hideaki*
Journal of Computational Chemistry, 26(8), p.788 - 798, 2005/06
Times Cited Count:24 Percentile:57.93(Chemistry, Multidisciplinary)Clustered DNA damage sites induced by ionizing radiation have been suggested to have serious consequences to organisms. In this study, approaches based on molecular dynamics (MD) simulation have been applied to examine conformational changes and energetic properties of DNA molecules containing clustered damage sites consisting of 2 lesioned sites, 8-oxoG and AP site. After 1 nanosecond of MD simulation, one of the 6 DNA molecules containing a clustered damage site develop specific characteristic features: sharp bending at the lesioned site and weakening or complete loss of electrostatic interaction energy between 8-oxoG and bases locating on the complementary strand. From these results it is suggested that these changes would make it difficult for the repair enzyme to bind to the lesions within the clustered damage site and thereby result in a reduction of its repair capacity.
Yonetani, Yoshiteru
Chemical Physics Letters, 406(1-3), p.49 - 53, 2005/04
Times Cited Count:52 Percentile:83.97(Chemistry, Physical)We report that a severe artifact appeared in molecular dynamics simulation of bulk water using the long cut-off length 18 AA ; Our result shows that increasing the cut-off length does not always improve the simulation result. Moreover, the use of the long cut-off length can lead to a spurious result. It is suggested that the simulation of solvated biomolecules using such a long cut-off length, which has been often performed, may contain an unexpected artifact.
Chikazumi, Shimpei*; Iwamoto, Akira
Chaos, Solitons and Fractals, 23(1), p.73 - 78, 2005/01
Times Cited Count:1 Percentile:11.41(Mathematics, Interdisciplinary Applications)To seek for a possible origin of fractal pattern in nature, we perform a molecular dynamics simulation for a fragmentation of an infinite fcc lattice. The fragmentation is induced by the initial condition of the model that the lattice particles have the Hubble-type radial expansion velocities. As time proceeds, the average density decreases and density fluctuation develops. By using the box counting method, it is found that the frequency-size plot of the density follows instantaneously a universal power-law for each Hubble constant up to the size of a cross-over. This cross-over size corresponds to the maximum size of fluctuation and is found to obey a dynamical scaling law as a function of time. This instantaneous generation of a nascent fractal is purely of dynamical origin and it shows us a new formation mechanism of a fractal patters different from the traditional criticality concept.
Kurosaki, Yuzuru; Yokoyama, Keiichi; Teranishi, Yoshiaki
Chemical Physics, 308(3), p.325 - 334, 2005/01
Times Cited Count:24 Percentile:60.84(Chemistry, Physical)A total of  1200 trajectories have been integrated for the two dissociation channels of formic acid, HCOOH HO + CO (1) and HCOOH CO + H (2), which occur with 248 and 193 nm photons, using the direct ab initio molecular dynamics method at the RMP2(full)/cc-pVDZ level of theory. It was found that the percentage of the energy distributed to a relative translational mode in reaction 2 is much larger than that in reaction 1. This is mainly due to the difference in the geometry of transition state (TS); the HO geometry in the TS of reaction 1 was predicted to significantly deviate from the equilibrium one, whereas the CO and H geometries in the TS of reaction 2 were found to be more similar to their equilibrium ones. It was also found that the product diatomic molecules, CO and H, are both vibrationally and rotationally excited. The calculated relative population of the vibrationally excited CO for the 248 nm photodissociation was consistent with experiment.
1200 trajectories have been integrated for the two dissociation channels of formic acid, HCOOH HO + CO (1) and HCOOH CO + H (2), which occur with 248 and 193 nm photons, using the direct ab initio molecular dynamics method at the RMP2(full)/cc-pVDZ level of theory. It was found that the percentage of the energy distributed to a relative translational mode in reaction 2 is much larger than that in reaction 1. This is mainly due to the difference in the geometry of transition state (TS); the HO geometry in the TS of reaction 1 was predicted to significantly deviate from the equilibrium one, whereas the CO and H geometries in the TS of reaction 2 were found to be more similar to their equilibrium ones. It was also found that the product diatomic molecules, CO and H, are both vibrationally and rotationally excited. The calculated relative population of the vibrationally excited CO for the 248 nm photodissociation was consistent with experiment.
Pinak, M.
JAERI 1346, 25 Pages, 2004/12
Molecular dynamics (MD) studies on several radiation damages to DNA - thymine dimer, thymine glycol8-oxoguanine and their recognition by repair enzymes are introduced in order to describe on the stepwise description of molecular process observed at radiation lesion sites. In all cases the significant structural changes in the DNA double helical structure were observed: (a) the breaking of hydrogen bonds network between complementary bases and resulting opening of the double helix (8-oxoguanine); (b) the sharp bending of the DNA helix centered at the lesion site (thymine dimer, thymine glycol); and (c) the flipping-out base on the strand complementary to the lesion (8-oxoguanine). The specific values of electrostatic interaction energy were found at the lesion sites (thymine dimmer: -10kcal/mol, thymine glycol: -26kcal/mol, 8-oxoguanine: -48kcal/mol).
Moritani, Kosuke; Okada, Michio*; Sato, Seiichi*; Goto, Seishiro*; Kasai, Toshio*; Yoshigoe, Akitaka; Teraoka, Yuden
Journal of Vacuum Science and Technology A, 22(4), p.1625 - 1630, 2004/08
Times Cited Count:25 Percentile:65.23(Materials Science, Coatings & Films)We studied the oxidation of Cu{111} surface with a hyperthermal O molecular beam (HOMB) using X-ray photoemission spectroscopy (XPS) in conjunction with a synchrotron radiation (SR) source. The efficiency of oxidation with 0.6-eV-HOMB is higher thab that with 2.3-eV-HOMB under 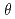
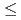 0.5ML. Ont the other hand, further oxidation occurs rather inefficiency under
0.5ML. Ont the other hand, further oxidation occurs rather inefficiency under 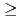 0.5ML. In this region, efficiency of oxidation with 2.3-eV-HOMB is higher than 0.6-eV-HOMB. We found that such slow oxidation process of Cu can be interpreted in terms of a collision-induced-adsorption mechanism. These results suggest that we can control the oxidation process of Cu by using HOMB.
0.5ML. In this region, efficiency of oxidation with 2.3-eV-HOMB is higher than 0.6-eV-HOMB. We found that such slow oxidation process of Cu can be interpreted in terms of a collision-induced-adsorption mechanism. These results suggest that we can control the oxidation process of Cu by using HOMB.
