


 CHO
CHO CH
CH +CO: Direct ab initio dynamics studyCHOCH+COの研究
+CO: Direct ab initio dynamics studyCHOCH+COの研究Kurosaki, Yuzuru; Yokoyama, Keiichi
RMP2(full)/cc-pVDZレベルでのdirect ab initioダイナミクス法を用い、S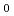 面上での光解離反応CHCHOCH+COのトラジェクトリを全部で100個計算した。相対並進エネルギー,CO内部エネルギー,CH内部エネルギーに対するエネルギー分配はそれぞれ、28,20,51%と計算された。生成物のCOは高回転励起状態に励起されるが、振動状態は励起されないことが予測され、平均の回転及び振動量子数はそれぞれ68.2,0.15と計算された。この結果は、Ghermanらの実験(J.Chem.Phys.2001, 114, 6128)と定性的に一致している。
面上での光解離反応CHCHOCH+COのトラジェクトリを全部で100個計算した。相対並進エネルギー,CO内部エネルギー,CH内部エネルギーに対するエネルギー分配はそれぞれ、28,20,51%と計算された。生成物のCOは高回転励起状態に励起されるが、振動状態は励起されないことが予測され、平均の回転及び振動量子数はそれぞれ68.2,0.15と計算された。この結果は、Ghermanらの実験(J.Chem.Phys.2001, 114, 6128)と定性的に一致している。
A total of 100 trajectories for the photodissociation, CHCHO CH + CO, on the S0 potential surface have been calculated using the direct ab initio molecular dynamics method at the RMP2(full)/cc-pVDZ level of theory. The energy distributions for the relative translational energy, the CO internal energy, and the CH internal energy were calculated to be 28, 20, and 51 %, respectively. It was predicted that the product CO is highly rotationally excited but vibrationally almost not excited; on average, the rotational and vibrational quantum numbers were 68.2 and 0.15, respectively, which qualitatively agrees with the recent observation of Gherman et al. (J. Chem. Phys. 2001, 114, 6128.)
発表言語 |
: | English |
|---|---|---|
掲載資料名 |
: | |
巻 |
: | 106 |
号 |
: | 47 |
ページ数 |
: | p.11415 - 11421 |
発行年月 |
: | 2002/11 |
キーワード |
: | Photodissociation; Acetaldehyde; Direct Ab Initio Dynamics; Product Energy Distribution |
論文URL |
: |
|
|---|---|---|
論文解説 |
: |
Access |
: |
- Accesses |
|---|---|---|
InCites™ |
: |
パーセンタイル:73.13 分野:Chemistry, Physical |
Altmetrics |
: |
[CLARIVATE ANALYTICS], [WEB OF SCIENCE], [HIGHLY CITED PAPER & CUP LOGO] and [HOT PAPER & FIRE LOGO] are trademarks of Clarivate Analytics, and/or its affiliated company or companies, and used herein by permission and/or license.


