


 O
O (001) surface; A DFT studyO (001)表面への水素の吸着に関する研究
(001) surface; A DFT studyO (001)表面への水素の吸着に関する研究Mao, W.*; 近田 拓未*; 志村 憲一郎*; 鈴木 晶大*; 山口 憲司; 寺井 隆幸*
Mao, W.*; Chikada, Takumi*; Shimura, Kenichiro*; Suzuki, Akihiro*; Yamaguchi, Kenji; Terai, Takayuki*
本研究では、密度汎関数理論に基づき、ErO(001)表面やその上に水素が吸着した状態を対象に、その構造的ならびに電子的特性に関する計算を実施した。計算の結果、非常に安定な吸着サイトがいくつか存在することが分かった。ErO(001)面上でエネルギー的に最も安定なサイトでは、水素の吸着エネルギーは295.68kJ mol (被覆率1/8ML)となり、この値は被覆率とともに減少する傾向にあった。さらに計算の結果、解離に伴う水素原子の吸着が、水素分子のそれと比べて、少なくとも吸着エネルギーが152.64kJ mol大きいことも分かった。これらの結果をもとに、核融合炉におけるトリチウム透過障壁中での水素同位体の透過挙動について考察した。
(被覆率1/8ML)となり、この値は被覆率とともに減少する傾向にあった。さらに計算の結果、解離に伴う水素原子の吸着が、水素分子のそれと比べて、少なくとも吸着エネルギーが152.64kJ mol大きいことも分かった。これらの結果をもとに、核融合炉におけるトリチウム透過障壁中での水素同位体の透過挙動について考察した。
In this work,  calculations based on density functional theory (DFT) and generalized gradient approximation were performed to investigate the structural and electronic properties of the cubic Er
calculations based on density functional theory (DFT) and generalized gradient approximation were performed to investigate the structural and electronic properties of the cubic Er O
O (001) surface and H adsorption processes on this surface. Several stable adsorption sites were identified, and at the most energetically favorable adsorption sites it was found that H bonds with O atoms at the cubic ErO (001) surface with an adsorption energy of 295.68 kJ mol at coverage 1/8 ML, which was inclined to decrease with the increase of H coverage (
(001) surface and H adsorption processes on this surface. Several stable adsorption sites were identified, and at the most energetically favorable adsorption sites it was found that H bonds with O atoms at the cubic ErO (001) surface with an adsorption energy of 295.68 kJ mol at coverage 1/8 ML, which was inclined to decrease with the increase of H coverage (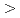 1/4 ML). In addition, the calculations revealed that the dissociative H atom configurations have adsorption energies that are at least 152.64 kJ mol greater than the H molecule configurations on the surface. These results are discussed in regard of the hydrogen isotope permeation behavior in the tritium permeation barrier in a fusion reactor.
1/4 ML). In addition, the calculations revealed that the dissociative H atom configurations have adsorption energies that are at least 152.64 kJ mol greater than the H molecule configurations on the surface. These results are discussed in regard of the hydrogen isotope permeation behavior in the tritium permeation barrier in a fusion reactor.
発表言語 |
: | English |
|---|---|---|
掲載資料名 |
: | |
巻 |
: | 443 |
号 |
: | 1-3 |
ページ数 |
: | p.555 - 561 |
発行年月 |
: | 2013/11 |
発表会議名 |
: | |
開催年月 |
: | 2012/10 |
開催都市 |
: | Osaka |
開催国 |
: | Japan |
キーワード |
: |
論文URL |
: |
|
|---|---|---|
論文解説 |
: |
Access |
: |
- Accesses |
|---|---|---|
InCites™ |
: |
パーセンタイル:30.44 分野:Materials Science, Multidisciplinary |
Altmetrics |
: |
[CLARIVATE ANALYTICS], [WEB OF SCIENCE], [HIGHLY CITED PAPER & CUP LOGO] and [HOT PAPER & FIRE LOGO] are trademarks of Clarivate Analytics, and/or its affiliated company or companies, and used herein by permission and/or license.
