


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
鈴木 健人*; 宮崎 貴暉*; 高柳 敏幸*; 志賀 基之
Physical Chemistry Chemical Physics, 20(41), p.26489 - 26499, 2018/11
被引用回数:2 パーセンタイル:6.30(Chemistry, Physical)ヘリウムクラスターの直接光イオン化に続く短時間過程について、DIMモデルに基づく経路積分分子動力学(PIMD)およびリングポリマー分子動力学(RPMD)シミュレーションで調べた。PIMDシミュレーションによって実験で得られた非対称で幅広いイオン化スペクトルが再現され、その原因はHe原子のエネルギー準位の不均一性にあることがわかった。また、RPMDシミュレーションから、高励起状態にあるイオン化ヘリウムクラスターは、非断熱的電荷移動を通じて高速電子状態緩和した後、ゆっくりと構造緩和することがわかった。
小座間 瑛記*; 安達 サディア*; 高柳 敏幸*; 志賀 基之
Chemistry; A European Journal, 24(48), p.12716 - 12721, 2018/08
被引用回数:5 パーセンタイル:17.57(Chemistry, Multidisciplinary)量子論的な経路積分分子動力学シミュレーションによって、ヘリウムクラスター中の三価アクチニウムカチオン(Ac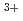 He
He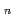 )の分子構造を計算した。安定なAc-He複合体が作られる低温(1-3K)において、ヘリウム原子の原子核量子効果は重要な役割を果たす。計算の結果、第一溶媒和圏を構成するヘリウム原子はD
)の分子構造を計算した。安定なAc-He複合体が作られる低温(1-3K)において、ヘリウム原子の原子核量子効果は重要な役割を果たす。計算の結果、第一溶媒和圏を構成するヘリウム原子はD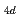 対称をもって配置され、その配位数は18個にも及ぶことがわかった。このAc-He
対称をもって配置され、その配位数は18個にも及ぶことがわかった。このAc-He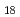 複合体は、クラスターサイズ(
複合体は、クラスターサイズ(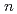 )が大きくなるにつれて強固になり、安定化する。なお、本研究は相対論的第一原理計算によりAc-He相互作用の正確な記述に基づくものである。
)が大きくなるにつれて強固になり、安定化する。なお、本研究は相対論的第一原理計算によりAc-He相互作用の正確な記述に基づくものである。
関 悠佑*; 高柳 敏幸*; 志賀 基之
Physical Chemistry Chemical Physics, 19(21), p.13798 - 13806, 2017/06
被引用回数:9 パーセンタイル:35.52(Chemistry, Physical)500個のヘリウム原子からなる低温クラスターに埋め込まれたAg原子の光励起ダイナミクスを理解するため、リングポリマー分子動力学シミュレーションを行った。本研究では、 Pスピン軌道の電子波動関数の時間依存シュレーディンガー方程式とRPMD方程式を結合したエーレンフェスト平均場近似を用いた。このシミュレーションから、Ag原子は光励起後100psの時間でヘリウムクラスターから大部分が放出されることが分かった。放出したAg原子の平均速度は60
Pスピン軌道の電子波動関数の時間依存シュレーディンガー方程式とRPMD方程式を結合したエーレンフェスト平均場近似を用いた。このシミュレーションから、Ag原子は光励起後100psの時間でヘリウムクラスターから大部分が放出されることが分かった。放出したAg原子の平均速度は60 70m/sと見積もられ、実験結果と定性的に一致した。
70m/sと見積もられ、実験結果と定性的に一致した。
箕島 裕介*; 関 悠佑*; 高柳 敏幸*; 志賀 基之
Chemical Physics, 472, p.1 - 8, 2016/06
被引用回数:2 パーセンタイル:5.44(Chemistry, Physical)放射線によって生じた電子がDNAに付着して生体損傷のもととなる際に分子構造がどのように変化するのか、基礎的問題として興味をもたれている。本研究では、グアニン-シトシン塩基対の電子付着過程について、経験的原子価結合モデルに基づく半古典リングポリマー分子動力学法を用いて理論的に解析した。その結果、塩基対の負イオンは、双極子束縛状態から原子価束縛状態へ短時間で変化し、その後塩基間のプロトン移動を引き起こすことがわかった。この過程における同位体置換効果や温度効果について詳しく調べた。プロトン移動の動的過程は、原子核の量子性を伴うものであることがわかった。
本田 知大*; 箕島 裕介*; 横井 悠輝*; 高柳 敏幸*; 志賀 基之
Chemical Physics Letters, 625, p.174 - 178, 2015/04
被引用回数:5 パーセンタイル:17.68(Chemistry, Physical)密度汎関数理論および分子動力学法により、グアニン-シトシン(G-C)塩基対への電子付着の動力学的ふるまいについて研究を行った。G-Cの負イオンのポテンシャルエネルギーは、長距離補正密度汎関数計算の情報をもとに経験的原子価結合法でモデル化した。半古典的リングポリマー分子動力学法の結果、初期において双極子束縛状態にある負イオンは、0.1ピコ秒程度で原子価束縛状態へと容易に移行した後、10ピコ秒程度でプロトン移動が起こることがわかった。一方、古典分子動力学法の結果でも同じ傾向が見られたが、反応にかかる時間はずっと遅かった。このことから、低エネルギー電子によるDNA損傷過程のダイナミクスにおいて、原子核の量子効果が重要な役割を果たしていると考えられる。
 -iron
-iron吉川 武宏*; 高柳 敏幸*; 君塚 肇*; 志賀 基之
Journal of Physical Chemistry C, 116(43), p.23113 - 23119, 2012/11
被引用回数:18 パーセンタイル:47.24(Chemistry, Physical)炉鋼材等の構造材料中を拡散する水素やその他の軽元素の挙動を知ることは、経年変化を評価するうえで重要な基礎的知見となり、計算機シミュレーションによる研究進展が期待されている。本発表では、分子シミュレーション法により、プロトンとトリチウムの鉄における拡散係数を100-1000Kの温度範囲で計算した成果を報告する。原子間相互作用にはEAMポテンシャルを利用し、近似的な量子動力学法であるセントロイド分子動力学法及びリングポリマー分子動力学法を用いて、量子トンネル効果を適切に考慮した。また、拡散経路について、経路積分分子動力学法に基づいた量子的遷移状態理論による解析を行った。プロトン拡散は500K、トリチウム拡散は300Kを境に熱的拡散から量子拡散へのクロスオーバーが見られた。トリチウムの拡散速度やそのメカニズムについては実験データが少ないため、このような分子シミュレーションによる予測は有効であると考えられる。
吉川 武宏*; 菅原 修一*; 高柳 敏幸*; 志賀 基之; 立川 仁典*
Chemical Physics, 394(1), p.46 - 51, 2012/02
被引用回数:18 パーセンタイル:53.69(Chemistry, Physical)プロトン移動反応は、水や生体内で起こる基本的反応であり、ポルフィセン分子は、それが協同的に起こる二重プロトン移動反応を示す典型的な系の一として多くの研究者の注目を受けている。本研究では、このポルフィセン分子における二重プロトン移動反応における同位体置換効果を調べるため、経路積分分子動力学シミュレーションを行った。その結果、室温において、ポルフィセンの二重プロトン移動は、おもに協同的機構で起こり、二次の鞍点の遷移状態を通過することで反応が進むことがわかった他、同位体置換は反応機構に大きな影響を与え、温度が上がると逐次反応の寄与が増えることも判明した。さらに、この際、面外の分子振動は、協同的機構を抑える働きをしていることもわかり、反応メカニズムの概略を理解することに成功した。これらの研究成果は、化学反応の基礎的理解を進めるものと位置付けられる一方、原子・分子のシミュレーション技術をより高精度にするための必須な技術開発である。
菅原 修一*; 吉川 武宏*; 高柳 敏幸*; 志賀 基之; 立川 仁典*
Journal of Physical Chemistry A, 115(42), p.11486 - 11494, 2011/09
被引用回数:20 パーセンタイル:56.27(Chemistry, Physical)本論文発表では、硫酸水和物クラスターにおけるイオン解離や水素結合交替プロセスを明らかにするため、量子的な経路積分分子動力学シミュレーションを行った成果について報告する。得られた成果は、クラスターサイズが大きくなるにつれ、硫酸の第1,第2脱プロトン化が起こりやすくなることがわかったほか、脱プロトン化過程の反応機構において、プロトン移動に関与する硫酸の酸素と水の酸素の間の距離が短くなるという知見である。これらの結果から、プロトンを受容する水の周りの配位数が重要な役割を担っていることが結論づけられる。本研究は、科学研究費補助金研究の一環として行われ、原子力材料物性シミュレーションの高度化手法を硫酸水和クラスターに応用することで得られた成果であり、基礎化学の発展に資する成果と位置付けられる。
吉川 武宏*; 菅原 修一*; 高柳 敏幸*; 志賀 基之; 立川 仁典*
Chemical Physics Letters, 496(1-3), p.14 - 19, 2010/08
被引用回数:28 パーセンタイル:67.10(Chemistry, Physical)全次元の経路積分分子動力学シミュレーションにより、ポルフィセンのプロトン移動による互変異性化が協奏的か逐次的かを調べた。その結果、二重プロトン移動は二次の鞍点構造を経る協奏的反応経路を通りやすいが、温度が高くなるにつれ、逐次的な機構の寄与が増すことがわかった。核の量子効果がプロトン移動メカニズムを決めるのに重要な役割を果たしている。
 O)
O)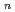 ( = 1-7) clusters on semi-empirical PM6 potential energy surfaces
( = 1-7) clusters on semi-empirical PM6 potential energy surfaces吉川 武宏*; 茂木 春樹*; 柿崎 陽*; 高柳 敏幸*; 志賀 基之
Chemical Physics, 365(1-2), p.60 - 68, 2009/12
被引用回数:15 パーセンタイル:44.94(Chemistry, Physical)水素結合性のglycine-(HO)(=1-7)クラスターについて、半経験的PM6ポテンシャルに基づいた経路積分分子動力学シミュレーションを行った。小さめの=1-3のクラスターについては、中性のグリシンと中性の水との間の水素結合錯体の構造をとる。一方、大きめの=4-6のクラスターでは、COOHカルボキシル基と水の間のプロトン移動が起こりやすくなるが、やはり全体の構造としては中性のグリシンと水である。これに対し、=7の場合は、グリシンがNH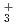 とCOO
とCOO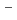 を持つ双性イオンの構造になりやすい。
を持つ双性イオンの構造になりやすい。
O)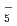 and (DO)
and (DO)高柳 敏幸*; 吉川 武宏*; 茂木 春樹*; 志賀 基之
Chemical Physics Letters, 482(4-6), p.195 - 200, 2009/12
被引用回数:10 パーセンタイル:28.12(Chemistry, Physical)水負イオンクラスター(HO) and (DO)について、半経験的1電子擬ポテンシャル分極モデルのもとで、経路積分分子動力学シミュレーションを行った。H原子の方がD原子より 零点振動の幅が大きいため、(HO)の水素結合距離は(DO)のものより少し長くなる。また、(HO)の垂直電子脱離エネルギーの分布幅は(DO)のそれよりも広くなることがわかった。このシミュレーションは、水負イオンクラスターにおける原子核の量子効果の重要性を示している。
SO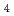
 (HO) (=1-6) on semiempirical PM6 potential surfaces
(HO) (=1-6) on semiempirical PM6 potential surfaces柿崎 陽*; 茂木 春樹*; 吉川 武宏*; 高柳 敏幸*; 志賀 基之; 立川 仁典*
Journal of Molecular Structure; THEOCHEM, 901(1-3), p.1 - 8, 2009/05
半経験的PM6法を用いて、一連の硫酸クラスターを対象とした量子的な経路積分分子動力学シミュレーションを行った。クラスターサイズが大きくなるほど、イオン解離を起こしやすくなり、コンタクトイオンペアと呼ばれる構造が支配的となる。クラスター構造やプロトン移動過程において原子核の量子効果が重要であることを示す。
O) on a semiempirical potential energy surface高柳 敏幸*; 高橋 健太*; 柿崎 陽*; 志賀 基之; 立川 仁典*
Chemical Physics, 358(3), p.196 - 202, 2009/04
被引用回数:16 パーセンタイル:47.46(Chemistry, Physical)塩酸クラスターHCl(HO)について、半経験的PM3-MAIS分子軌道計算から基底状態のポテンシャル面を求め、経路積分分子動力学シミュレーションを行った。計算の結果、300K以上の温度では構造変化を起こし、液体的な振るまいが見られた。この構造変化では、水素原子核の量子揺らぎが重要な役割を担っている。
茂木 春樹*; 柿崎 陽*; 高柳 敏幸*; 武次 ゆり子*; 武次 徹也*; 志賀 基之
Chemical Physics, 354(1-3), p.38 - 43, 2008/12
被引用回数:12 パーセンタイル:38.05(Chemistry, Physical)He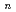 BeOクラスターにおける量子的なヘリウムの溶媒和構造を明らかにするため、経路積分分子動力学シミュレーションを行った。その結果1個のヘリウム原子がBeO分子と強く結合し、その周りを12-14からなる第一溶媒和圏のヘリウム原子が取り囲む構造が明らかとなった。この計算はHENDI法による実験においてヘリウムクラスター中にHeBeO錯体が見つかる可能性を示唆している。
BeOクラスターにおける量子的なヘリウムの溶媒和構造を明らかにするため、経路積分分子動力学シミュレーションを行った。その結果1個のヘリウム原子がBeO分子と強く結合し、その周りを12-14からなる第一溶媒和圏のヘリウム原子が取り囲む構造が明らかとなった。この計算はHENDI法による実験においてヘリウムクラスター中にHeBeO錯体が見つかる可能性を示唆している。
 O) (=2-7) clusters on semiempirical PM6 potential energy surfaces
O) (=2-7) clusters on semiempirical PM6 potential energy surfaces高柳 敏幸*; 吉川 武宏*; 柿崎 陽*; 志賀 基之; 立川 仁典*
Journal of Molecular Structure; THEOCHEM, 869(1-3), p.29 - 36, 2008/11
半経験的分子軌道法であるPM6法に基づいた分子動力学法により、グリシン-水クラスターの熱力学的構造を調べた。クラスターサイズが大きくなるにつれグリシンが中性分子であるよりも双性イオンであるほうが安定になる傾向が見られた。また、中性分子と双性イオンの形での水和構造の違いについて議論する。
system黒崎 譲; 高柳 敏幸*
Chemical Physics Letters, 406(1-3), p.121 - 125, 2005/04
BrH系の非経験的に計算された基底状態のグローバルなポテンシャルエネルギー面(PES)についての新しい解析的関数を構築した。これは、以前われわれが発表した1A' PES[Y. Kurosaki, T. Takayanagi, J. Chem. Phys. 119 (2003) 7383]の修正版である。反応H+HBr H+Brとその同位体置換した反応の速度定数を、新しい1A' PESを用いて計算したところ実測値をよく再現した。これはフィットした関数の反応障壁の値1.53kcal mol
H+Brとその同位体置換した反応の速度定数を、新しい1A' PESを用いて計算したところ実測値をよく再現した。これはフィットした関数の反応障壁の値1.53kcal mol が真の値に非常に近いことを強く示唆している。
が真の値に非常に近いことを強く示唆している。
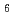
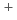 in irradiated solid
in irradiated solid  -hydrogen and its decay dynamics; Reinvestigation of quartet electron paramagnetic resonance lines assigned to H
-hydrogen and its decay dynamics; Reinvestigation of quartet electron paramagnetic resonance lines assigned to H熊田 高之; 田地川 浩人*; 高柳 敏幸*
Physical Chemistry Chemical Physics, 7(5), p.776 - 784, 2005/02
被引用回数:12 パーセンタイル:37.60(Chemistry, Physical)以前Hと同定されていた放射線照射固体パラ水素中に観測された4本線の電子スピン共鳴(ESR)信号を再調査した。実験と理論計算値を照らし合わせた結果、4本線はHではなくHのものであることが判明した。新しい同定のもとに、以前に測定されたオルト-パラ変換,同位体効果,量子拡散などのデータを全て解析し直した。本論文の最後には、気体水素の放射線分解ではH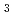 イオンが生じるのとは対照的に、固体水素に放射線を照射したときに生成するイオン種はHであるというモデル提案した。
イオンが生じるのとは対照的に、固体水素に放射線を照射したときに生成するイオン種はHであるというモデル提案した。
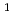 D)+NONO+NO/N+O reaction
D)+NONO+NO/N+O reaction高柳 敏幸
Chemical Physics, 308(3), p.211 - 216, 2005/01
被引用回数:6 パーセンタイル:19.94(Chemistry, Physical)O(D)+NO反応について量子-古典波束法を用いた理論計算を行った。計算は平面対称性を仮定した5次元で行い、NO分子の3振動自由度を量子波束法によって取り扱い、残りの2自由度を古典力学で取り扱った。以前われわれが開発した高精度分子軌道法の計算結果をもとにして開発した解析的なポテンシャルエネルギー曲面を用いた。この計算の目的は2つの反応生成チャンネル、NO+NO及びN+Oが衝突エネルギーやNO分子の初期振動量子状態によってどのように変化するかを理論的に調べることである。計算の結果、衝突エネルギーの増加とともに、NO+NOの生成確率が減少し、N+Oチャンネルが逆に増加することを見いだした。一方、生成分岐比はNOの初期振動量子状態によってほとんど影響を受けないことがわかった。これらの計算結果は、成層圏での熱非平衡下で起こるO(D)+NO反応のメカニズムを理解するうえで極めて重要である。
高柳 敏幸
Chemical Physics, 302(1-3), p.85 - 93, 2004/07
被引用回数:12 パーセンタイル:36.10(Chemistry, Physical)量子-古典混合近似を用いてアセトニトリルクラスターへの電子付加ダイナミクスの理論シミュレーションを行った。過剰電子の運動は電子基底状態を保つよう、量子力学的に取り扱い、溶媒分子中の原子の運動は古典力学で取り扱った。一電子擬ポテンシャルを電子とアセトニトリル分子の相互作用に使い、溶媒分子間の相互作用については経験的なポテンシャルを用いた。初期のクラスター構造は一定温度の分子動力学計算によって求めた。過剰電子は非常に弱く束縛された表面電子の状態から、徐々に溶媒和され、数ピコ秒の後、溶媒和される。このとき、アセトニトリル中のメチル基が電子に向くように分子が回転する。低温での小さなクラスターを除いて、ほぼすべてのクラスターで内部に束縛された溶媒和電子が生成することが見いだされた。電子の束縛エネルギーを最近の実験結果と比較したところ、シミュレーションは実験値よりかなり大きくなることがわかった。これは使った擬ポテンシャルにさらに改良の余地があることを示している。
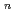 exciplexes
exciplexes高柳 敏幸; 志賀 基之
Physical Chemistry Chemical Physics, 6(13), p.3241 - 3247, 2004/07
被引用回数:30 パーセンタイル:68.11(Chemistry, Physical)300個のヘリウム原子からなるクラスターに付着したカリウム原子の光吸収ダイナミクスを最近われわれが開発した非断熱ハイブリッド量子シミュレーション法を用いて理論的に調べた。この方法では、カリウム原子のP電子を量子的に取り扱い、ヘリウム原子の運動は半古典経路積分セントロイド分子動力学法によって取り扱う。数十個のトラジェクトリーを計算した結果、初期励起の種類やクラスター初期構造に依存せず、すべての場合でカリウム原子がクラスターから脱離することがわかった。K*Heエキシマーの生成メカニズムを調べるため、ダイナミクスが断熱ポテンシャル面上で起こると仮定した計算も行い、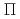 型の電子励起を行った場合にエキシマーが生成することを見いだした。これらの計算結果は過去の実験結果と定性的に一致する。また、エキシマー中のヘリウムの溶媒和構造を経路積分分子動力学法によって調べ、第一励起状態では6個のヘリウム原子が第一溶媒和相に束縛されるのに対し、第二励起状態では2個だけが束縛されることがわかった。
型の電子励起を行った場合にエキシマーが生成することを見いだした。これらの計算結果は過去の実験結果と定性的に一致する。また、エキシマー中のヘリウムの溶媒和構造を経路積分分子動力学法によって調べ、第一励起状態では6個のヘリウム原子が第一溶媒和相に束縛されるのに対し、第二励起状態では2個だけが束縛されることがわかった。
