


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
高見澤 悠; 清水 康雄*; 井上 耕治*; 野沢 康子*; 外山 健*; 矢野 史子*; 井上 真雄*; 西田 彰男*; 永井 康介*
Applied Physics Express, 9(10), p.106601_1 - 106601_4, 2016/10
被引用回数:0 パーセンタイル:0(Physics, Applied)The effect of phosphorus (P) and boron (B) doping on arsenic (As) diffusion in polycrystalline silicon (poly-Si) was investigated using laser-assisted atom probe tomography. In all samples, a high concentration of As was found at the grain boundaries, indicating that such boundaries represent the main diffusion path. However, As grain-boundary diffusion was suppressed in the B-doped sample, and enhanced in the P-doped sample. In a sample co-doped with both P and B, As diffusion was somewhat enhanced, indicating competition between the effects of the two dopants. This can be explained by the pairing of P and B atoms. The results suggest that grain-boundary diffusion of As can be controlled by varying the local concentration of P and B dopants.
石田 恒
Proteins: Structure, Function, and Bioinformatics, 82(9), p.1985 - 1999, 2014/09
被引用回数:10 パーセンタイル:32.43(Biochemistry & Molecular Biology)Proteasome is involved in the degradation of proteins. Proteasome activators bind to the proteasome core particle (CP) and facilitate opening a gate of the CP, where Tyr8 and Asp9 in the N-termini tails of the CP form the ordered open gate. Four different molecular dynamics simulations were carried out: ordered- and Tyr8Gly/Asp9Gly disordered-gate models of the CP complexed with an ATP-independent PA26 and ordered- and disordered-gate models of the CP complexed with an ATP-dependent PAN-like activator. In the ordered-gate models, the substrate in the activator was more stable than that in the CP. In the disordered-gate models, the substrate in the activator was more destabilized than in the ordered-gate models. Thus, it was concluded that the dynamics of the N-termini tails entropically play a key role in the translocation of the substrate.
石田 恒; 松本 淳
PLOS ONE (Internet), 9(7), p.e101951_1 - e101951_17, 2014/07
被引用回数:16 パーセンタイル:44.05(Multidisciplinary Sciences)To understand the mechanism of tRNA translocation in ribosome, all-atom molecular dynamics simulations of reverse translocation were carried out. Ribosome-tRNAs-mRNA-EFG complex at the post-translocational state was directed towards the translocational and pre-translocational states by fitting the complex into cryo-EM density maps. Multistep structural changes, such as a ratchet-like motion between the small and large ribosomal subunits and rotation of the head of the small subunit were observed during translocation. onformational change of EF-G was interpreted as the result of the combination of the internal and external motions: the internal hinge-bending motion around axes from one subunit to the other, and external rotational motion triggered by L12 around an axis passing near the sarcin-ricin loop. These motions contributed to the extension of domain IV of EF-G to maintain its interaction with the A/P-tRNA during tRNA translocation.
岸田 潔*; 細田 尚*; 澤田 淳; 佐藤 久; 中島 伸一郎*; 安原 英明*
Harmonising Rock Engineering and the Environment, p.1327 - 1330, 2011/10
自然の亀裂中の開口部は不均質な分布を呈するにもかかわらず、一般に均質な平行平板モデルで表現され、亀裂の透水特性の評価には三乗則が適用される。平行平板モデルで三乗則が適用されるのは、レイノルズ数が1.0以下であり、移流項が流れに影響しないことが知られている。本研究では、レイノルズ数が1以下の条件下での単一亀裂透水実験に対し、移流項を考慮したモデル(2Dモデル)による2次元浸透流解析を行った。得られた結果から、局所レイノルズ数の評価と三乗則成立に加えて2Dモデルの検証に関する検討を行った。
石田 恒
Progress in Nuclear Science and Technology (Internet), 2, p.470 - 476, 2011/10
We have developed an integrated molecular simulation system for biological macromolecules, called SCUBA (Simulation Codes for hUge Biomolecular Assembly) which is designed to run a biomolecule system composed of more than one million particles efficiently on parallel computers. SCUBA is now being used not only for molecular dynamics (MD) simulations but also to determine the 3D structure of supra-macromolecules. For the latter purpose, a new method to fit a high-resolution X-ray structure at a certain reaction state into low-resolution electron microscopy (EM) data at different reaction states has been developed. In our method, the fitting is carried out using MD simulations in explicit water medium; the target function considers restraints to fit the X-ray structure into an EM density map, and the all-atom interactions for all the molecules including the water medium. This method was implemented into SCUBA and applied to ribosome, one of the supra-biomolecules utilized for translating genetic information to the amino acid sequence. The system was composed of more than 2 million atoms. The method showed that SCUBA can carry out the fitting simulation with a high parallelization efficiency ratio of more than 50% using 512 CPU cores of PRIMERGY BX900 supercomputer at the Japan Atomic Energy Agency.
石田 恒
Journal of Computational Chemistry, 31(12), p.2317 - 2329, 2010/09
Branch migration of the Holliday junction takes place at the center of the RuvA tetramer. To elucidate how branch migration occurs, umbrella sampling simulations were performed for complexes of the RuvA tetramer and Holliday junction DNA. While conventional umbrella sampling simulations set sampling points a priori, the umbrella sampling simulation in this study set the sampling points one by one in order to search for a realistic path of the branch migration during the simulations. Starting from the X-ray structure of the complex, in which the hydrogen bonds between two base-pairs were unformed, the hydrogen bonds between the next base-pairs of the shrinking stems were observed to start to disconnect. At the intermediate stage, three or four of the eight unpaired bases interacted closely with the acidic pins from RuvA. During the final stage, these bases moved away from the pins and formed the hydrogen bonds of the new base-pairs of the growing stems. The free-energy profile along this reaction path showed that the intermediate stage was ameta-stable state between two free-energy barriers of about 10-15 kcal/mol. These results imply that the pins play an important role in stabilizing the interactions between the pins and the unpaired base-pairs.
岸田 潔*; 澤田 淳; 佐藤 久; 音田 慎一郎*; 細田 尚*
第39回岩盤力学に関するシンポジウム講演論文集(CD-ROM), p.287 - 292, 2010/01
自然の亀裂は、複雑な開口幅分布を有することが知られているが、透水挙動を評価する場合、一般には平行平板モデルに代表される等価な亀裂開口幅として扱い、三乗則が適用される。平行平板モデルで三乗則が適用されるのは、レイノルズ数が1以下の極めて遅い流れであり、移流項が流れに影響しないとされている。本研究では、レイノルズ数が1以下の三乗則が成立する条件下での単一亀裂透水実験に対し、慣性項を考慮したモデルによる2次元浸透流解析を行った。得られた結果から、局所レイノルズ数を算定し、三乗側成立条件下での単一亀裂透水実験における亀裂内の局所三乗則の成立性に関する検討を行った。
松本 淳; 石田 恒
Structure, 17(12), p.1605 - 1613, 2009/12
被引用回数:20 パーセンタイル:48.25(Biochemistry & Molecular Biology)Many three-dimensional density maps of 70S ribosome at various functional states are available now in the Electron Microscopy DataBank at EBI. We used our new flexible-fitting approach to systematically analyze these maps to reveal the global conformational differences between the EM structures. Large-scale ratchet-like deformations were observed in an EM structure of the initiation complex and in some EM structures bound with EFG, RF3, and RRF. In most of them, the L1 stalk, which interacts with the tRNA molecule at the E site of ribosome and is considered to be involved in the release of the tRNA, was in "the blocking state" for the E-tRNA. Furthermore, we found that the EM structures bound with EFG or RRF were aligned in the conformational space, suggesting that the large-scale conformational changes of the 70S ribosome bound with these factors occur along a specific pathway in a concerted manner.
石田 恒; 河野 秀俊
Research Advances in Nucleic Acids Research, p.19 - 34, 2009/03
Ribosome is the macromolecular machine composed of RNA and protein used in the expression of the genetic code for the synthesis of polypeptides. The polypeptide is generated at the peptidyl transferase center and then passes through a tunnel in ribosome. It is known that some nascent polypeptides specifically interact with the tunnel in ribosome and regulate the function of ribosome. The tunnel is also thought to play an active role in the passage of the nascent polypeptides through the tunnel. In this review, we summarise both experimental and theoretical research on the regulation of the nascent polypeptide in the ribosomal tunnel and the role of the tunnel in the passage of the polypeptide.
石田 恒; Hayward, S.*
Biophysical Journal, 95(12), p.5962 - 5973, 2008/12
被引用回数:29 パーセンタイル:60.62(Biophysics)Molecular dynamics simulations were performed on  70S ribosome with and without the nascent polypeptide inside the exit tunnel. Modeling of the polypeptide in the tunnel revealed two possible paths: one over Arg
70S ribosome with and without the nascent polypeptide inside the exit tunnel. Modeling of the polypeptide in the tunnel revealed two possible paths: one over Arg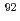 of L22 and one under (from the viewpoint of 50S on top of 30S). A strong interaction between L4 and Arg was observed without the polypeptide and when it passed over Arg. However, when the polypeptide passed under, Arg repositioned to interact with Ade
of L22 and one under (from the viewpoint of 50S on top of 30S). A strong interaction between L4 and Arg was observed without the polypeptide and when it passed over Arg. However, when the polypeptide passed under, Arg repositioned to interact with Ade of 23S rRNA. Using steered molecular dynamics the polypeptide could be pulled through the L4-L22 constriction when situated under Arg, but did not move when over. These results suggest that the tunnel is closed by the Arg-L4 interaction before elongation of the polypeptide and the tunnel leads the entering polypeptide from the peptidyl transferase center to the passage under Arg, causing Arg to switch to an open position. It is possible, therefore, that Arg plays the role of a gate, opening and closing the tunnel at L4-L22. There is controversy over whether the tunnel is dynamics or rigid. At least within the time-scale of our simulations conformational analysis showed that global motions mainly involve relative movement of the 50S and 30S subunits and appear not to affect the conformation of the tunnel.
of 23S rRNA. Using steered molecular dynamics the polypeptide could be pulled through the L4-L22 constriction when situated under Arg, but did not move when over. These results suggest that the tunnel is closed by the Arg-L4 interaction before elongation of the polypeptide and the tunnel leads the entering polypeptide from the peptidyl transferase center to the passage under Arg, causing Arg to switch to an open position. It is possible, therefore, that Arg plays the role of a gate, opening and closing the tunnel at L4-L22. There is controversy over whether the tunnel is dynamics or rigid. At least within the time-scale of our simulations conformational analysis showed that global motions mainly involve relative movement of the 50S and 30S subunits and appear not to affect the conformation of the tunnel.
Poornam, G. P.*; 松本 淳; 石田 恒; Hayward, S.*
Proteins: Structure, Function, and Bioinformatics, 76(1), p.201 - 212, 2008/12
被引用回数:83 パーセンタイル:90.94(Biochemistry & Molecular Biology)A new method for the analysis of domain movements in large, multichain, biomolecular complexes is presented. The method is applicable to any molecule for which two atomic structures are available that represent a conformational change indicating a possible domain movement. The method is blind to atomic bonding and atom type and can, therefore, be applied to biomolecular complexes containing different constituent molecules such as protein, RNA, or DNA. Here, we report on the application of the method to biomolecules covering a considerable size range: hemoglobin, liver alcohol dehydrogenase, S-Adenosylhomocysteine hydrolase, aspartate transcarbamylase, and the 70S ribosome. The results provide a depiction of the conformational change within each molecule that is easily understood, giving a perspective that is expected to lead to new insights.
 transition edge sensor
transition edge sensor岡安 悟; 片桐 政樹; 北條 喜一; 森井 幸生; 三木 重信*; 島影 久志*; Wang, Z.*; 石田 武和*
Physica C, 468(15-20), p.1998 - 2000, 2008/09
被引用回数:0 パーセンタイル:0(Physics, Applied)MgB超伝導体の超伝導転移端(TES)を利用した中性子センサーの開発を行っている。この目的のため低ノイズの測定系を開発してきた。この測定系の有用性を検証するため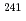 Amを用いた
Amを用いた 線検出を試みた。感度をかせぐため試料は1
線検出を試みた。感度をかせぐため試料は1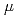 m幅で35mの長さの短いミアンダパターンを用いた。定電流モードで測定を行ったところ線の検出に成功した。バイアス電流は6Aであった。
m幅で35mの長さの短いミアンダパターンを用いた。定電流モードで測定を行ったところ線の検出に成功した。バイアス電流は6Aであった。
 thin film detector for neutrons
thin film detector for neutrons石田 武和*; 西川 正利*; 藤田 賢文*; 岡安 悟; 片桐 政樹*; 佐藤 和郎*; 四谷 任*; 島影 久志*; 三木 茂人*; Wang, Z.*; et al.
Journal of Low Temperature Physics, 151(3-4), p.1074 - 1079, 2008/05
被引用回数:36 パーセンタイル:78.05(Physics, Applied)本論文では、ボロン同位体(質量数10)を増量した超伝導MgB中性子検出器は比較的高い温度で操作可能であることを示す。基本動作原理は、ボロン同位体が中性子をよく吸収し、核反応を起こすことで、超伝導転移近傍で大きな電気抵抗変化が瞬間的に起こることであり、実験用の原子炉から射出される冷中性子が高感度で検出可能となる。出力となる発生電位差については、デジタルオシロスコープを用いて低ノイズの増幅装置を用いることで十分に検出可能であることが分かった。また、詳細な上記核反応により起こる超伝導非平衡ダイナミクスについては、時間依存のギンツブルク・ランダウ方程式のシミュレーションをスーパーコンピュータ上で実施することにより追跡可能であり、観測事実とよく符号することが分かっている。
石田 恒; 由良 敬; 叶野 琢磨; 松本 淳
Annual Report of the Earth Simulator Center April 2006 - March 2007, p.257 - 263, 2007/09
地球シミュレータは従来にはない大規模生体超分子系の分子動力学シミュレーションを可能とする計算能力を持つ。われわれは生体超分子系を扱う大規模な分子動力学シミュレーションシステムSCUBAを開発している。SCUBAは、地球シミュレータ360プロセッサ使用時でベクトル化率95%以上,並列化効率50%以上の優れた性能を達成している。本年度はSCUBAが必要とするメモリ使用量を最適化するためのチューニングをすることにより、百万原子以上からなる生体超分子の分子動力学シミュレーションを地球シミュレータ上で実行可能とした。さらに、マルチ時間ステップ法を装備することにより長時間シミュレーションを実行可能とした。そして、約200万原子からなるリボソーム(遺伝情報を翻訳する生体超分子)と新生ポリペプチドの複合体の系について分子動力学シミュレーションを実行した。結果、リボソームの機能に重要と考えられているねじれ運動を再現することに成功した。
石田 恒; 樋口 真理子; 米谷 佳晃*; 叶野 琢磨; 城地 保昌*; 北尾 彰朗*; 郷 信広
Annual Report of the Earth Simulator Center April 2005 - March 2006, p.237 - 240, 2007/01
地球シミュレータは従来にはない大規模生体超分子系の分子動力学シミュレーションを可能とする計算能力を持つ。そこで、われわれは数百万原子の生体超分子系を扱う大規模な分子動力学シミュレーションシステムSCUBA(旧名PABIOS)を開発している。SCUBAはPPM計算法,系のエネルギー,温度,圧力を一定に保つさまざまな時間積分アルゴリズム,系の原子分布異方性により引き起こされる並列化効率の悪化を防ぐための動的ロードバランスなど、最新のアルゴリズムを採用した計算性能に優れたシミュレーションシステムである。本年度は、約55万原子の系(RuvAB-Holliday分岐DNA)について分子動力学シミュレーションを実行した結果、地球シミュレータで360個のプロセッサを用いてもSCUBAはベクトル化率95%以上,並列化効率50%以上の優れた性能を達成することに成功した。そして、ホリデイジャンクションモデル(RuvA-Holliday分岐DNA)の長時間分子動力学シミュレーションを実行することで、Holliday分岐DNA結合タンパク質RuvAがDNAを組み換えるメカニズムを分子レベルで明らかにすることができた。
石田 恒; 松本 淳; 堤 遊*; 由良 敬
Proceedings of 16th International Microscopy Congress (IMC 2006), P. 242, 2006/09
生体超分子の電子顕微鏡像を取得することによって、いろいろな状態での構造を知ることができる。本研究では蛋白質合成生体超分子であるリボゾームの構造変化を電子顕微鏡像と計算科学の手法によって推定した。
石田 恒; 樋口 真理子; 米谷 佳晃*; 叶野 琢磨; 城地 保昌*; 北尾 彰朗*; 郷 信広
Annual Report of the Earth Simulator Center April 2004 - March 2005, p.241 - 246, 2005/12
地球シミュレータは従来にはない大規模超分子系の分子動力学シミュレーションを可能とする計算能力を持つ。そこで、われわれは数百万原子のシステムを扱う大規模な分子シミュレーション(PABIOS)を開発している。PABIOSは空間分割法を用いており高い並列化効率を持つ。PABIOSは系のエネルギー,温度,圧力を一定に保つさまざまな時間積分アルゴリズム,原子結合長を固定することにより時間ステップの増大を可能とするSHAKE, RATTLEアルゴリズムなどの高精度アルゴリズムを採用した計算性能に優れたシミュレーションシステムである。今回、系の原子分布の異方性による並列化効率の悪化がおこらないように、動的ロードバランスを開発した。ホリデイ分岐DNAとDNA結合蛋白質RuvA4量体の複合体が水中に存在する系(全系16万6千原子)について分子動力学シミュレーションを実行した結果、地球シミュレータで120個のプロセッサを用いてもPABIOSはベクトル化率95%以上,並列化効率50%以上の優れた性能を達成することに成功した。そして計算速度も昨年度と比べて約2倍程度向上した。
石田 恒; 城地 保昌*; 樋口 真理子; 叶野 琢磨; 北尾 彰朗*; 郷 信広
Annual Report of the Earth Simulator Center April 2003 - March 2004, p.175 - 179, 2004/07
地球シミュレータは従来にはない大規模超分子系の分子動力学シミュレーションを可能とする計算能力を持つ。そこで、われわれは数百万原子のシステムを扱う大規模な分子シミュレーション(PABIOS)を開発している。PABIOSは空間分割法を用いており高い並列化効率を持つ。PABIOSは系のエネルギー,温度,圧力を一定に保つさまざまな時間積分アルゴリズム,原子結合長を固定することにより時間ステップの増大を可能とするSHAKE, RATTLEアルゴリズムなどの高精度アルゴリズムを採用した計算性能に優れたシミュレーションシステムである。ホリデイ分岐DNAとDNA結合蛋白質RuvA4量体の複合体が水中に存在する系(全系16万6千原子)について分子動力学シミュレーションを実行した結果、地球シミュレータで144個のプロセッサを用いてもPABIOSはベクトル化率95%以上,並列化効率50%以上の優れた性能を達成することに成功した。
逆井 章; 石田 真一; 松川 誠; 秋野 昇; 安藤 俊就*; 新井 貴; 江里 幸一郎; 濱田 一弥; 市毛 尚志; 礒野 高明; et al.
Nuclear Fusion, 44(2), p.329 - 334, 2004/02
超伝導トカマク装置へのJT-60改修が計画されている。原型炉に繋がる先進的な核融合技術として、JT-60改修装置(JT-60SC)の設計のために超伝導マグネット技術やプラズマ対向機器を開発した。JT-60SCの超伝導トロイダル磁場コイル用として、高い臨界電流密度を可能とする、高い銅比4のニオブアルミ超伝導素線を新規に開発し、量産化に成功した。この素線と、突合せ溶接で作った全長30mの丸穴四角のステンレス製コンジットを用いて、実機サイズのケーブル・イン・コンジット導体を製作した。この導体を使用して、リアクト&ワインド法(熱処理後に巻線作業を行う製作方法)を実証するR&Dを進めている。ニオブアルミ導体の歪み劣化が小さいことを利用したこの製作方法は、将来の大型コイル製作の技術的な信頼性向上と低コストに繋がる先進的な超伝導技術として注目されている。JT-60SCのダイバータへの熱負荷10-15MW/m に耐える機器として、スクリュウ管を銅製ヒートシンクに設け、これと炭素繊維複合材,緩衝材を一体ロウ付けすることで、良好なプラズマ対向機器を開発した。電子ビーム照射試験により、この対向機器は従来のスワール管の場合と比較して約1.5倍の高い熱伝達率を達成することを明らかにした。
に耐える機器として、スクリュウ管を銅製ヒートシンクに設け、これと炭素繊維複合材,緩衝材を一体ロウ付けすることで、良好なプラズマ対向機器を開発した。電子ビーム照射試験により、この対向機器は従来のスワール管の場合と比較して約1.5倍の高い熱伝達率を達成することを明らかにした。
逆井 章; 石田 真一; 松川 誠; 秋野 昇; 安藤 俊就*; 新井 貴; 江里 幸一郎; 濱田 一弥; 市毛 尚志; 礒野 高明; et al.
Nuclear Fusion, 44(2), p.329 - 334, 2004/02
被引用回数:7 パーセンタイル:22.95(Physics, Fluids & Plasmas)超伝導トカマク装置へのJT-60改修が計画されている。原型炉に繋がる先進的な核融合技術として、JT-60改修装置(JT-60SC)の設計のために超伝導マグネット技術やプラズマ対向機器を開発した。JT-60SCの超伝導トロイダル磁場コイル用として、高い臨界電流密度を可能とする、高い銅比4のニオブアルミ超伝導素線を新規に開発し、量産化に成功した。この素線と、突合せ溶接で作った全長30 mの丸穴四角のステンレス製コンジットを用いて、実機サイズのケーブル・イン・コンジット導体を製作した。この導体を用いて、リアクト&ワインド法(熱処理後に巻線作業を行う製作方法)を実証するR&Dを進めた。ニオブアルミ導体の歪み劣化が小さいことを利用したこの製作方法は、将来の大型コイル製作の技術的な信頼性向上と低コストに繋がる先進的な超伝導技術として注目されている。JT-60SCのダイバータへの熱負荷10-15MW/mに耐える機器として、スクリュウ管を銅製ヒートシンクに設け、これと炭素繊維複合材、緩衝材を一体ロウ付けすることで、良好なプラズマ対向機器を開発した。電子ビーム照射試験により、この対向機器は従来のスワール管の場合と比較して約1.5倍の高い熱伝達率を達成することを明らかにした。
