


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
 path-integral simulations of hydrogen-isotope diffusion in face-centred cubic metals
path-integral simulations of hydrogen-isotope diffusion in face-centred cubic metals君塚 肇*; 尾方 成信*; Thomsen, B.; 志賀 基之
Journal of Physics; Condensed Matter, 37(19), p.193001_1 - 193001_16, 2025/04
被引用回数:0 パーセンタイル:0.00(Physics, Condensed Matter)第一原理経路積分法を用いて、面心立方格子金属の水素同位体拡散係数を計算したところ、拡散係数の温度依存性が特異な形状をとることがわかった。これは温度依存性の異なる核量子効果の競合に起因しており、通常の同位体効果と逆同位体効果の間の異常なクロスオーバーのメカニズムを説明するものである。また、密度汎関数理論計算とほぼ同じ精度で、機械学習ベースの原子間ポテンシャルを用いて、近似的な量子ダイナミクスを記述する経路積分シミュレーションの結果も紹介する。この計算法は、様々な固体物質における水素同位体の量子的振る舞いを解明する道を開くものである。
志賀 基之; Thomsen, B.; 君塚 肇*
Physical Review B, 109(5), p.054303_1 - 054303_12, 2024/02
被引用回数:3 パーセンタイル:74.90(Materials Science, Multidisciplinary)パラジウム中の水素の中性子非弾性散乱スペクトルを有限温度での核量子効果を考慮して計算した。経路積分に基づく半古典分子動力学と機械学習ポテンシャルを組合せた計算手法を用いた。計算されたスペクトルは、水素原子の振動励起の基本波と第一高調波に対応するピークの位置と強度に関して、実験スペクトルとよく一致した。古典分子動力学法との比較から、中性子非弾性散乱スペクトルにおいて核量子効果が本質的な役割を果たしていることが示された。
Kwon, H.*; 志賀 基之; 君塚 肇*; 小田 卓司*
Acta Materialia, 247, p.118739_1 - 118739_11, 2023/04
被引用回数:21 パーセンタイル:94.41(Materials Science, Multidisciplinary)機械学習によるモーメントテンソルポテンシャルを用いた経路積分シミュレーションから、体心立方格子金属(Nb, Fe, W)中の希薄水素の拡散係数を密度汎関数理論レベルの精度で推定した。この計算結果は、精度が高いと考えられるいくつかの実験結果と大いに一致した。また、実験結果と矛盾なく同位体効果を再現した。
君塚 肇*; Thomsen, B.; 志賀 基之
Journal of Physics; Energy (Internet), 4(3), p.034004_1 - 034004_13, 2022/07
被引用回数:18 パーセンタイル:78.16(Chemistry, Physical)パラジウム-水素系の人工ニューラルネットワークに基づく原子間ポテンシャルを開発するとともに、パラジウム結晶中の水素同位体の量子拡散について、経路積分分子動力学計算を行った。50-1500Kの広い温度範囲で軽水素と重水素の拡散係数を計算し、低温と高温における拡散機構の違いを明らかにした。
 path integral simulations
path integral simulations君塚 肇*; 志賀 基之
Physical Review Materials (Internet), 5(6), p.065406_1 - 065406_9, 2021/06
被引用回数:14 パーセンタイル:58.48(Materials Science, Multidisciplinary)金属内部における水素の動的挙動において、核量子効果は無視できない因子である。本研究では、核量子効果を考慮した第一原理積分分子動力学シミュレーションを用いて、面心立方金属Al, Ag, Cuにおける水素拡散を調べた。その結果、AgとCuにおける水素拡散の温度依存性は逆S字型の曲線となるのに対して、Alにおける水素拡散の温度依存性はC字型となることがわかった。この違いは、水素が最安定となる位置がAg, Cuでは八面体サイト、Alでは四面体サイトであることに由来する。このことから、水素拡散の核量子効果(零点振動とトンネリング)は安定サイトの異なる金属によって質的に異なることがわかった。
君塚 肇*; 尾方 成信*; 志賀 基之
日本物理学会誌, 75(8), p.484 - 490, 2020/08
水素は量子性を示す元素で、他の不純物原子にはない特異な拡散挙動を示す。本研究では、経路積分シミュレーションと電子状態計算を連成させた第一原理手法を用いて、パラジウム結晶中の水素同位体の拡散挙動を幅広い温度で予測した。その結果、高温領域では、温度の低下とともに量子ゆらぎの影響が顕在化することによって拡散抑制が生じ、アレニウスプロットが上に凸の折れ曲がることが示された。一方、低温域では、量子トンネル効果の発現によって拡散障壁は減少に転じ、アレニウスプロットは下に凸の折れ曲がることが示された。異なる温度領域における量子効果の競合は、水素拡散の特異な同位体効果を明瞭に説明する。
君塚 肇*; 尾方 成信*; 志賀 基之
Physical Review B, 100(2), p.024104_1 - 024104_9, 2019/07
被引用回数:20 パーセンタイル:66.51(Materials Science, Multidisciplinary)結晶中の水素同位体の振る舞いは材料物理学における基本的なテーマである。広い温度範囲にわたる同位体拡散挙動は重要な知見だが、まだ完全な調査には至っていない。本研究では、電子と原子核の両方を量子力学的に扱った最先端の第一原理計算を行い、面心立方Pd金属中の水素同位体拡散係数の温度依存性を調べたところ、逆S型形状のアレニウスプロットを示すことがわかった。この異常な振る舞いは、温度・質量依存性の異なる核量子効果が競合することに起因する。その結果、80-400Kの限られた温度範囲で、三重水素(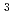 H)が軽水素(
H)が軽水素(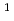 H)よりも速く拡散するという特性につながる。このメカニズムは、高温および低温での実験でそれぞれ観察されている正・逆同位体効果間がクロスオーバーを示す理論的根拠となることがわかった。
H)よりも速く拡散するという特性につながる。このメカニズムは、高温および低温での実験でそれぞれ観察されている正・逆同位体効果間がクロスオーバーを示す理論的根拠となることがわかった。
君塚 肇*; 尾方 成信*; 志賀 基之
Physical Review B, 97(1), p.014102_1 - 014102_11, 2018/01
被引用回数:33 パーセンタイル:77.82(Materials Science, Multidisciplinary)パラジウム(Pd)中の水素(H)の高い拡散性のメカニズムを理解するため、有限歪み下における電子と核の両方の量子性を考慮した第一原理シミュレーションに基づいて、面心立方晶PdにおけるHの格子間拡散プロセスを研究した。その結果、八面体サイトの核量子効果のために、温度の低下とともに水素移動の活性化障壁が大幅に増加することが明らかになった。しかしながら、拡大した格子歪みのもとでは、四面体サイトがより安定化し、核量子効果はあまり顕著にならなかった。これは、歪みが増加するにつれて、拡散機構が量子的拡散から古典的拡散へ徐々に変化することを意味する。
 -iron
-iron吉川 武宏*; 高柳 敏幸*; 君塚 肇*; 志賀 基之
Journal of Physical Chemistry C, 116(43), p.23113 - 23119, 2012/11
被引用回数:18 パーセンタイル:47.24(Chemistry, Physical)炉鋼材等の構造材料中を拡散する水素やその他の軽元素の挙動を知ることは、経年変化を評価するうえで重要な基礎的知見となり、計算機シミュレーションによる研究進展が期待されている。本発表では、分子シミュレーション法により、プロトンとトリチウムの鉄における拡散係数を100-1000Kの温度範囲で計算した成果を報告する。原子間相互作用にはEAMポテンシャルを利用し、近似的な量子動力学法であるセントロイド分子動力学法及びリングポリマー分子動力学法を用いて、量子トンネル効果を適切に考慮した。また、拡散経路について、経路積分分子動力学法に基づいた量子的遷移状態理論による解析を行った。プロトン拡散は500K、トリチウム拡散は300Kを境に熱的拡散から量子拡散へのクロスオーバーが見られた。トリチウムの拡散速度やそのメカニズムについては実験データが少ないため、このような分子シミュレーションによる予測は有効であると考えられる。
蕪木 英雄; Li, J.*; Yip, S.*; 君塚 肇*
Journal of Applied Physics, 102(4), p.043514_1 - 043514_6, 2007/08
被引用回数:46 パーセンタイル:81.26(Physics, Applied)グリーン-久保の線形応答理論に基づいた平衡分子動力学法を用いて希ガス結晶アルゴンの熱伝導率を計算した。原子間相互作用の計算には、カットオフ距離を十分に長く取ったレナード・ジョーンズポテンシャルを用いた。この方法により、固体から液体までの熱伝導率の絶対値を予測するとともに、熱流束の相関関数の時間依存性から熱伝導の動的過程について考察した。その結果、低温において相関関数が2つの緩和過程から構成されていることを示すとともに、短時間の緩和が局所的な単一粒子運動、長時間の緩和が原子の集団運動であるフォノンに対応していることを示した。温度が上昇するに従い長時間の相関は短時間の相関に比較して速く減少することを見いだし、これが固体において位相にコヒーレントなフォノン輸送から位相にインコヒーレントな原子間エネルギーへの遷移に対応していることを示した。
清水 大志; 尾方 成信*; 君塚 肇*; 叶野 琢磨; Li, J.*; 蕪木 英雄
Journal of the Earth Simulator, 7, p.17 - 21, 2007/06
Predicting atomistic properties of a dislocation is a first step toward an understanding of plastic behavior of materials, in particular BCC metals. The core structure and Peierls stress of a screw dislocation in BCC metals have been studied over the years using the first-principles and empirical methods, however, their conclusions vary due to the inefficiency of the methods. We have executed first-principles calculations based on the density functional method, employing the most accurate 1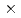 120 k-point samplings, to determine the core structure and Peierls stress of the
120 k-point samplings, to determine the core structure and Peierls stress of the 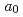 /2[111] screw dislocation of molybdenum. We have concluded that the core has a 6-fold structure, and determined the Peierls stress of 1.8 GPa for the simple shear strain along the (
/2[111] screw dislocation of molybdenum. We have concluded that the core has a 6-fold structure, and determined the Peierls stress of 1.8 GPa for the simple shear strain along the (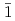 10)
10) 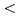 111
111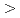 direction.
direction.
門吉 朋子; 蕪木 英雄; 清水 大志; 君塚 肇*; 實川 資朗; Li, J.*
Acta Materialia, 55(9), p.3073 - 3080, 2007/05
被引用回数:72 パーセンタイル:92.16(Materials Science, Multidisciplinary)分子動力学法により、FCC金属中の積層欠陥正四面体が、不等辺六角形転位ループから形成されることを確かめた。また、格子間原子型のフランクループ及び原子空孔型のフランクループが、外部せん断応力や温度によって完全転位ループに遷移する過程を詳細に解析し、完全転位化に温度が重要な役割を果たしていることを明らかにした。
清水 大志; 君塚 肇*; 蕪木 英雄; 荒川 忠一*
計算工学講演会論文集, 7(1), p.163 - 166, 2002/05
分子動力学法は現実の物質系をミクロなレベルからシミュレーションする手法であり、ニュートンの運動方程式を直接決定論的に数値積分することにより、系の静的な特性とともに動的な特性を求めることができる。分子動力学法シミュレーションにおいて最も計算時間を要する部分は一般的に原子間の相互作用の計算であり、大規模なシミュレーションを行なうには相互作用のカットオフにより計算コストを減らす高速化手法を用いることが必要である。また、今日では計算の高速化や大容量のメモリを確保するのに並列計算が有効である。われわれは、並列化やカットオフによる高速化などのシミュレーション手法を記述するプログラムの機能単位を切り分け、これらを再構成することにより並列分子動力学ステンシルを開発した。並列分子動力学ステンシルを利用すると、並列化手法及びカットオフによる高速化手法について意識することなくプログラミングを行なうことが可能である。本論文では、並列分子動力学ステンシルの設計及び性能について報告する。
清水 大志; 君塚 肇*; 蕪木 英雄; 荒川 忠一*
日本計算工学会論文集, 4, p.225 - 230, 2002/04
分子動力学法(MD)シミュレーションにおいて最も計算時間を要する部分は一般的に原子間の相互作用の計算である。各原子に作用する力を単純なアルゴリズムで計算するとペアを検索するコストが対象とする系の原子数の2乗に比例することから、短距離力の系について大規模なシミュレーションを行なう際は、粒子間に作用する力にカットオフを設定して計算コストを減らすカットオフ手法を用いることが必須となっている。また、計算の高速化や大容量のメモリを確保するのに並列計算が有効である。われわれは並列化などのシミュレーション手法の記述と系の性質の記述が分離した、見通しの良いプログラムを作成する枠組みとして並列分子動力学ステンシルを開発した。並列分子動力学ステンシルは、原子間相互作用を計算するプログラムを対象となる系の性質(モデル)の記述のみとなることを実現している。
板倉 憲一; 横川 三津夫; 清水 大志; 君塚 肇*; 蕪木 英雄
情報処理学会研究報告2001-HPC-88, p.67 - 72, 2001/10
地球シミュレータは、640の計算ノードを持ち理論ピーク性能は40Tflop/sである。プロセッサノードはピーク性能8Gflop/sのベクトルプロセッサ8個,16GBの共有メモリから構成される。本研究では地球シミュレータの計算ノードによる固体分子動力学法の計算プログラムのベクトル化と並列化を行い性能評価を行った。分子動力学法では、カットオフ半径内の粒子が互いに影響を与え、その粒子ペアを行列を用いて表現することができる。ベクトル化に際して、この行列表現にcompressed row formとjagged diagonalformを考える。jagged diagonal formはベクトル長がcompressedrow formよりも長くできるので、ベクトル化により適している。しかし、標準的な粒子対の情報からjagged diagonal formに変換するには時間がかかるため、全体の性能はより簡単なcompressedrow fromよりも低下した。compressed row formでは8CPUでの並列化により2.4から2.7倍のスピードアップとなった。
山田 進; 清水 大志; 今井 隆太*; 君塚 肇*; 加治 芳行; 蕪木 英雄
計算工学講演会論文集, 6(1), p.233 - 236, 2001/05
日本原子力研究所では科学技術計算に比較的多くあらわれる基本的な数値計算である連立一次方程式の反復法,固有値問題,フーリエ変換,擬似乱数生成の各ルーチンについて、メッセージパッシングを用いた並列計算ルーチンを開発している。この並列計算ルーチン群は並列数値計算ライブラリPARCEL(Parallel Computing Elements)として公開されており、実際に数多くの国内外の大学等の研究機関で利用されている。本研究では、PARCELの連立一次方程式の反復解法を用いてベクトル並列計算機上で構造解析を行い、その結果からデータの格納法や前処理等についての並列性能を評価した。
清水 大志; 君塚 肇*; 蕪木 英雄; Li, J.*; Yip, S.*
Proceedings of 4th International Conference on Supercomputing in Nuclear Applications (SNA 2000) (CD-ROM), 10 Pages, 2000/09
並列分子動力学(MD)について、さまざまな並列化手法やカットオフを用いた計算の効率化手法を組み合わせたシミュレーションをプログラムするのは簡単ではない。われわれは大規模な並列MDシミュレーションのためのプログラムの基本パターンである「並列分子動力学ステンシル」を開発した。本ステンシルは並列化手法や効率化手法を分離し、プログラム作成者がシミュレーション自体に専念できるように設計されている。プログラムの全体はC言語とMPIを用いるが、各MPI関数の呼び出しはプログラム作成者から隠蔽されている。本論文では並列MDステンシルを用い15~75Kの固体アルゴン(結晶状態及びアモルファス状態)に関して500~1,000,000原子を用いたシミュレーションを行い、その弾性について研究した。
-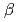 transition of cristobalite, SiO
transition of cristobalite, SiO ; A Molecular-dynamics study
; A Molecular-dynamics study君塚 肇*; 蕪木 英雄; 木暮 嘉明*
Physical Review Letters, 84(24), p.5548 - 5551, 2000/06
被引用回数:145 パーセンタイル:94.60(Physics, Multidisciplinary)特異な弾性的性質を示すことで知られる及びクリストバライトについて、300~1800Kの温度範囲に渡る断熱弾性スティフネス定数を、平衡分子動力学法によりゆらぎの式を用いて求めた。クリストバライトはこの幅広い温度領域において負のポアソン比を示すが、相と相ではその機構は異なる。さらに立方結晶である相においては、コーシーの関係に反して弾性定数C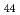 がC
がC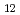 よりもむしろC
よりもむしろC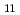 と極めて近い値を示すことが確認された。このことは[100]方向に伝播する弾性波の縦波、横波の速度が一致するという特筆すべき性質を表す。
と極めて近い値を示すことが確認された。このことは[100]方向に伝播する弾性波の縦波、横波の速度が一致するという特筆すべき性質を表す。
山田 進; 清水 大志; 小林 謙一*; 君塚 肇*; 岸田 則生*; 蕪木 英雄
計算工学講演会論文集, p.369 - 372, 2000/05
日本原子力研究所で開発した並列数値計算ライブラリ「PARCEL」を紹介する。このライブラリは係数行列が疎行列であるような連立一次方程式の反復解法、実対称及びエルミート行列の固有値の計算、模擬一様乱数の生成ルーチン及び高速フーリエ変換のルーチンから構成されている。また、PARCELの各ルーチンはFORTRAN77及びMPIを用いて記述されており、さまざまな計算機上で利用できる特長を持っている。さらに、演算の分割や通信と計算のオーバーラップなどの工夫により優れたスケーラビリティを持つようにプログラムされている。そこで、このルーチンのスケーラビリティを調べるため、実際に並列計算機を用いて実行した結果、優れたスケーラビリティを持つことが確認できた。
清水 大志; 君塚 肇*; 蕪木 英雄
計算工学講演会論文集, p.361 - 364, 2000/05
大規模数値シミュレーションの分野において並列計算は必須の手法となっているが、並列計算機ではプロセッサ間のデータ転送の記述等、逐次計算機では見られなかったプログラム作成の難しさがある。物質、材料、生体などの分野において需要の高い大規模分子動力学法の並列化においてはシミュレーションの計算手法と並列化の手法が密接にかかわるように書かれ、非常に複雑になってしまうことが多い。並列分子動力学ステンシルは、これらを分離することで、見通しの良いシミュレーションプログラムを作成する枠組みを提供するものである。また、この枠組みに沿ってプログラミングを行う際に利用可能な部品をライブラリとして用意した。

