


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
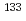 CsI +
CsI + 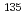 Cs
Cs  Cs + ICs
Cs + ICs小林 孝徳*; 松岡 雷士*; 横山 啓一
Computational and Theoretical Chemistry, 1150, p.40 - 48, 2019/02
被引用回数:1 パーセンタイル:1.58(Chemistry, Physical)セシウムの同位体分離法の開発に関連して、ヨウ化セシウム分子とセシウム原子の衝突によるセシウム交換反応の反応速度定数を量子化学計算及び擬古典トラジェクトリー計算により評価した。その結果、3.6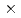 10
10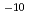 cm
cm /molecule/sという大きな値が得られた。また、わずかながら正の温度依存性を持つことが示され、長距離相互作用による引力ポテンシャルと解離プロセスの影響がその原因と考えられた。
/molecule/sという大きな値が得られた。また、わずかながら正の温度依存性を持つことが示され、長距離相互作用による引力ポテンシャルと解離プロセスの影響がその原因と考えられた。
 )
)黒崎 譲; 横山 啓一
Computational and Theoretical Chemistry, 999, p.239 - 245, 2012/11
被引用回数:5 パーセンタイル:10.06(Chemistry, Physical)Potential energy curves (PECs) for the low-lying states of the diatomic cesium iodide cation (CsI) have been calculated using the internally-contracted MRSDCI method with relativistic pseudpotentials. First, we calculate PECs for spin-orbit (SO)-free 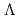 -S states,
-S states, 
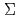 and
and 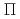 , stemming from the triply degenerate P state of I. Then we obtain PECs for SO-included
, stemming from the triply degenerate P state of I. Then we obtain PECs for SO-included 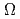 states, X3/2, 1/2(I), and 1/2(II), by diagonalizing the matrix of the electronic Hamiltonian plus SO coupling. It is found that all of the three state PECs, X3/2, 1/2(I), and 1/2(II), have shallow wells with the depths of 1.4-4.6 kcal mol
states, X3/2, 1/2(I), and 1/2(II), by diagonalizing the matrix of the electronic Hamiltonian plus SO coupling. It is found that all of the three state PECs, X3/2, 1/2(I), and 1/2(II), have shallow wells with the depths of 1.4-4.6 kcal mol . It is also predicted that transition dipole moments (TDMs) among these states have values of non-negligible amounts around equilibrium internuclear distances and the TDMs can contribute to the optical transition between the states.
. It is also predicted that transition dipole moments (TDMs) among these states have values of non-negligible amounts around equilibrium internuclear distances and the TDMs can contribute to the optical transition between the states.
 path integral Monte Carlo simulations for water trimer with electron correlation effects
path integral Monte Carlo simulations for water trimer with electron correlation effects藤田 貴敏*; 田中 成典*; 藤原 崇幸*; 草 将晃*; 望月 祐志*; 志賀 基之
Computational and Theoretical Chemistry, 997, p.7 - 13, 2012/10
被引用回数:9 パーセンタイル:19.95(Chemistry, Physical)固体や液体等において、低原子量の元素から構成される化合物のダイナミクスを解析する場合には、原子核の量子ダイナミクスを考慮する必要が指摘されており、原子力分野をはじめとしてさまざまな分野において、それを考慮した新規のシミュレーション手法の開発等が議論されてきている。本発表では、この観点に立ち、電子と原子核からなる系をまるごと量子力学的に扱った第一原理経路積分モンテカルロシミュレーションにより、三量体の水クラスターの構造を精密に調べた結果を報告する。なお、採用した手法は、三次の多体摂動論に基づいた電子状態理論で求められた断熱ポテンシャル上のもとで、量子及び熱的効果を含めた分子構造の動的なゆらぎを求めることのできる極めて精度の高いシミュレーション手法であり、計算の結果、水クラスターの水素結合の柔軟性は、従来の第一原理計算から予測されていた結果よりも大きく、酸素間距離が大きな振幅で振動する様子が明らかになった。
path integral Monte Carlo simulations for water trimer with electron correlation effects藤田 貴敏*; 田中 成典*; 藤原 崇幸*; 草 将晃*; 望月 祐志*; 志賀 基之
Computational and Theoretical Chemistry, 997, p.7 - 13, 2012/10
水の三量体とは、凝集相にある水分子間の水素結合状態を特徴づけるモデルとして、実験と理論の両面から活発に研究が行われている系である。本論文では、この水三量体を対象として、電子相関効果を三次のM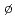 ller-Plesset摂動法まで考慮した高精度の第一原理経路積分モンテカルロ法を用いて、電子と原子核からなる系をすべて量子的に扱った第一原理シミュレーションを行った。具体的には、水三量体におけるO-O距離, O-H-O角度, H-OOO二面角など、水素結合を特徴づける構造因子を対象として、核の量子効果,熱的ゆらぎ効果,電子相関効果が与える影響について、系統だった解析を行い、これにより、計算で得られた水素結合構造の誤差を定量的に見積ることが可能となり、実験データとの直接比較ができるようになった。これらの成果は、水分子間の水素結合の基礎的理解を進めるものだが、高精度な原子分子レベルの計算手法の開発とその検証に該当し、原子力分野での応用が期待される。
ller-Plesset摂動法まで考慮した高精度の第一原理経路積分モンテカルロ法を用いて、電子と原子核からなる系をすべて量子的に扱った第一原理シミュレーションを行った。具体的には、水三量体におけるO-O距離, O-H-O角度, H-OOO二面角など、水素結合を特徴づける構造因子を対象として、核の量子効果,熱的ゆらぎ効果,電子相関効果が与える影響について、系統だった解析を行い、これにより、計算で得られた水素結合構造の誤差を定量的に見積ることが可能となり、実験データとの直接比較ができるようになった。これらの成果は、水分子間の水素結合の基礎的理解を進めるものだが、高精度な原子分子レベルの計算手法の開発とその検証に該当し、原子力分野での応用が期待される。
杉本 昌崇*; 志賀 基之; 立川 仁典*
Computational and Theoretical Chemistry, 975(1-3), p.31 - 37, 2011/11
被引用回数:5 パーセンタイル:10.66(Chemistry, Physical)本論文では、原子核の量子統計的ゆらぎを考慮した第一原理分子動力学シミュレーションを用いて、一連の水素クラスター陽イオンH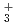 , H
, H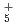 , H
, H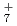 , H
, H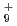 の解析を行った。その結果,クラスターの解離反応H
の解析を行った。その結果,クラスターの解離反応H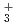 (H
(H )
)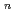 H(H)
H(H)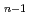 +Hに必要なエネルギーが5.4, 2.1, 3.2kcal/molと見積もられ、実験値と定性的に一致した結果が得られた。解析の結果、水素クラスター解離過程は、量子的な零点振動とその非調和性の影響を強く受けることがわかった。以上、本計算手法を用いると、同位体を有する化合物の化学反応における同位体効果が計算可能であり、原子力材料シミュレーション研究の高度化に資する成果である。
+Hに必要なエネルギーが5.4, 2.1, 3.2kcal/molと見積もられ、実験値と定性的に一致した結果が得られた。解析の結果、水素クラスター解離過程は、量子的な零点振動とその非調和性の影響を強く受けることがわかった。以上、本計算手法を用いると、同位体を有する化合物の化学反応における同位体効果が計算可能であり、原子力材料シミュレーション研究の高度化に資する成果である。
黒崎 譲; 横山 啓一; 横山 淳
Computational and Theoretical Chemistry, 963(2-3), p.245 - 255, 2011/02
被引用回数:6 パーセンタイル:13.14(Chemistry, Physical)We study, using quantum optimal control theory (OCT), isotope-selective vibrational excitations on the ground-state potential energy curve for the mixture of two pure ensembles for CsI and CsI. Initial states are set to the condition that both CsI and CsI are in the vibrational ground level, i.e., (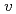
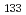 ,
, 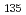 ) = (0, 0), and target states are set to three: (, ) = (0, 2), (0, 3), and (0, 4), where molecular orientations are fixed parallel to the field polarization vector. Three total times (i.e., pulse durations), T = 460,000 au, 920,000 au, and 1,840,000 au, are set in the calculations and nine cases for the combination of target state and total time are investigated. It is suggested from the computational results that even when T is short, high-yield transitions into energetically-separated target states are possible through excitations skipping over more than one vibrational level at a time with intense fields including high overtones.
) = (0, 0), and target states are set to three: (, ) = (0, 2), (0, 3), and (0, 4), where molecular orientations are fixed parallel to the field polarization vector. Three total times (i.e., pulse durations), T = 460,000 au, 920,000 au, and 1,840,000 au, are set in the calculations and nine cases for the combination of target state and total time are investigated. It is suggested from the computational results that even when T is short, high-yield transitions into energetically-separated target states are possible through excitations skipping over more than one vibrational level at a time with intense fields including high overtones.
