


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
甲斐 健師; 樋川 智洋; 松谷 悠佑*; 平田 悠歩; 土田 秀次*; 横谷 明徳*
Journal of Chemical Physics, 162(15), p.154102_1 - 154102_11, 2025/04
被引用回数:0 パーセンタイル:0.00(Chemistry, Physical)放射線DNA損傷を推定するには、水の放射線分解の結果生じる低エネルギー電子の科学的知見が必要となる。しかしながら、水の放射線分解の解析は非常に複雑であるため本研究では、シンプルな水の光分解に関する低エネルギー電子の実験値と、水中の電極への光照射により発生した低エネルギー電子の実験値に注目した。本研究ではモンテカルロ法と分子動力学法を組み合わせた計算コードを利用し、これらの実験値を解析した。その結果、異なる実験条件であっても実験値をよく再現することを確認した。本計算コードは低エネルギー電子とDNAの相互作用を解析する強力なツールとなり、放射線DNA損傷の形成メカニズムの解明に適用されることが期待される。
 and machine learning potentials for modeling nuclear quantum effects in water
and machine learning potentials for modeling nuclear quantum effects in waterThomsen, B.; 永井 佑紀*; 小林 恵太; 濱田 幾太郎*; 志賀 基之
Journal of Chemical Physics, 161(20), p.204109_1 - 204109_18, 2024/11
被引用回数:1 パーセンタイル:37.13(Chemistry, Physical)SL-PIHMC-MIX法は、第一原理計算と機械学習ポテンシャルを混合した自己学習型経路積分ハイブリッドモンテカルロ法である。SL-PIHMC-MIX法を用いると、第一原理経路積分分子動力学法(PIMD法)よりも、室温の水の構造を計算し収束させるのに必要な第一原理DFT計算の回数を一桁減らすことができる。
土田 秀次*; 手塚 智哉*; 甲斐 健師; 松谷 悠佑*; 間嶋 拓也*; 斉藤 学*
Journal of Chemical Physics, 161(10), p.104503_1 - 104503_8, 2024/09
被引用回数:0 パーセンタイル:0.00(Chemistry, Physical)高速イオンビームは、生体細胞内の水との相互作用によって生成される二次電子などの化学生成物によってDNAに損傷を与えるが、粒子線治療で用いられるブラッグピーク領域におけるこれらの化学生成物の生成過程は完全には理解されていない。この過程を調べるために、真空中の液体水ジェットにMeVエネルギーの炭素ビームを照射したときに生成される放射線分解物の収率を評価する実験を行った。さらに、放射線輸送モンテカルロコードを用いて、入射イオンと二次電子による水中の電離過程をシミュレーションした。その結果、水中でのイオン化の主な原因は二次電子であることがわかった。最後に、これらの素過程は、DNA損傷の形成機構を研究する放射線生物物理学や生化学の発展に寄与することを示す。
樋川 智洋; 甲斐 健師; 熊谷 友多; 横谷 明徳*
Journal of Chemical Physics, 160(21), p.214119_1 - 214119_9, 2024/06
被引用回数:3 パーセンタイル:75.15(Chemistry, Physical)イオン化によって引き起こされる不均質反応であるスパー反応は、溶液中の放射線分解あるいは光分解反応を左右する重要な反応だが、そのスパーの形成プロセスはまだ解明されていない。その理由の1つとして、イオン化によって生成した荷電種を取り囲む溶媒和分子の誘電応答の影響がまだ明らかになっていないことが挙げられる。誘電応答は誘電率の時間変化に対応しており、スパー形成プロセスにおける反応拡散系に影響を与える可能性がある。そこで本研究では、誘電応答を考慮しながらDebye-Smoluchowski方程式を解くことにより、反応拡散系に対する誘電応答の影響を調べた。荷電種間に働くクーロン力は、誘電応答とともに徐々に減少する。本計算から、誘電応答が完了する前に荷電種間で反応が起こる条件を見積もることが出来た。これまで低LET放射線誘起によるイオン化で生成する自由電子の初期G値が静的な誘電率に依存することは報告されているが、荷電種間が密になる高LET放射線や光誘起の化学反応を扱う場合は誘電応答を考慮することが重要であることが示唆された。
 He spin filter and small-angle X-ray scattering
He spin filter and small-angle X-ray scattering領木 研之*; 渡部 史*; 奥平 琢也*; 高橋 慎吾*; 奥 隆之; 廣井 孝介; 元川 竜平; 中村 洋*
Journal of Chemical Physics, 160(11), p.114907_1 - 114907_9, 2024/03
被引用回数:2 パーセンタイル:37.13(Chemistry, Physical)Small-angle neutron scattering (SANS) and small-angle X-ray scattering (SAXS) measurements were performed for deuterated and non-deuterated poly(ethylene glycol) (d-PEG and h-PEG, respectively) in D O and a DO/HO mixed solvent (Mix) to compere the scattering profiles. To determine the coherent scattering intensity of SANS, a He spin filter was utilized. The scattering profiles determined by SANS measurements were analyzed in terms of the wormlike chain model with touched beads along the contour of the chain. However, the SAXS profiles were not explained by the same model with uniform beads but with beads each consisting of a core and a shell having different electron densities. To explore the chain thickness determined form the SANS profile, scattering intensities for different combinations of d-PEG/DO, d-PEG/Mix, h-PEG/DO, and h-PEG/Mix were also examined.
O and a DO/HO mixed solvent (Mix) to compere the scattering profiles. To determine the coherent scattering intensity of SANS, a He spin filter was utilized. The scattering profiles determined by SANS measurements were analyzed in terms of the wormlike chain model with touched beads along the contour of the chain. However, the SAXS profiles were not explained by the same model with uniform beads but with beads each consisting of a core and a shell having different electron densities. To explore the chain thickness determined form the SANS profile, scattering intensities for different combinations of d-PEG/DO, d-PEG/Mix, h-PEG/DO, and h-PEG/Mix were also examined.
越田 裕之*; Wilde, M.*; 福谷 克之
Journal of Chemical Physics, 160(3), p.034703_1 - 034703_9, 2024/01
被引用回数:0 パーセンタイル:0.00(Chemistry, Physical)We have developed an experimental and analytical setup for thermal desorption spectroscopy of solid water films on surfaces. We obtain the coverage-dependent desorption kinetics of water molecules from a well-defined ultra-thin alumina/NiAl(110) surface in the coverage range of 0-2 monolayers. We use a novel deconvolution technique to eliminate the pumping delay of water vapor in the vacuum system, which has previously hindered the accurate estimation of desorption kinetic parameters, such as activation energy and pre-exponential factor. The coverage-dependent Arrhenius analysis reveals that the desorption activation energy decreases with increasing coverage in the submonolayer range, indicating that the water-water interaction is not attractive. We also find that the pre-exponential factor for the second layer is higher than that for the sub-monolayer. We explain this difference in terms of transition state theory and propose that entropic effects play a significant role in water desorption kinetics.
甲斐 健師; 樋川 智洋; 鵜飼 正敏*; 藤井 健太郎*; 渡邊 立子*; 横谷 明徳*
Journal of Chemical Physics, 158(16), p.164103_1 - 164103_8, 2023/04
被引用回数:6 パーセンタイル:69.12(Chemistry, Physical)水の放射線分解・光分解に関する新たな科学的知見は、放射線化学・放射線生物学を含む様々な研究分野の劇的進歩に必要不可欠である。水に放射線を照射すると、その飛跡上に沿って、反応性の高い水和電子が無数に生成される。水和電子は、発生した電子と水分子の運動が動的に相関し、形成されることは知られているが、その形成に至るまでの、電子の非局在化、熱化、分極メカニズムは未だ解明していない。本研究で独自に開発したコードを利用した解析結果から、これらの過渡的現象は、水特有の水素結合ネットワークに由来する分子間振動モードと、水和を進行する水分子の回転モードの時間発展に支配されるように進行することが明らかとなった。本研究によるアプローチは、水に限らず、様々な溶媒に適用可能であり、そこから得られる科学的知見は、放射線生物影響、原子力化学、放射線計測など幅広い研究領域へ適用されることが期待できる。
species at SiO/Si interfaces in Si dry oxidation; Comparison between p-Si(001) and n-Si(001) surfaces津田 泰孝; 吉越 章隆; 小川 修一*; 坂本 徹哉*; 山本 善貴*; 山本 幸男*; 高桑 雄二*
Journal of Chemical Physics, 157(23), p.234705_1 - 234705_21, 2022/12
被引用回数:2 パーセンタイル:14.89(Chemistry, Physical)This paper gives experimental evidence for that (1) the excess minority carrier recombination at the SiO /p-Si(001) and SiO/n-Si(001) interfaces is associated with the O dissociative adsorption, (2) the 700-eV X-ray induced enhancement of the SiO growth is not caused by the band flattening due to the surface photovoltaic effect but ascribed to the electron-hole pair creation due to core level photoexcitation for the spillover of the bulk Si electronic states to the SiO layer, (3) changes of band bending result from the excess minority carrier recombination at the oxidation-induced vacancy site when turning on and off the X-ray irradiation, and (4) a metastable chemisorbed O species (Pb1-paul) plays a decisive role in combining two kinds of the reaction loops of single- and double-step oxidation. Based on the experimental results, the unified Si oxidation reaction model mediated by point defect generation [Jpn. J. Appl. Phys. 59, SM0801 (2020)] is extended from a viewpoint of (a) the excess minority carrier recombination at the oxidation-induced vacancy site and (b) the trapping-mediated adsorption through the chemisorbed O species at the SiO/Si interface.
/p-Si(001) and SiO/n-Si(001) interfaces is associated with the O dissociative adsorption, (2) the 700-eV X-ray induced enhancement of the SiO growth is not caused by the band flattening due to the surface photovoltaic effect but ascribed to the electron-hole pair creation due to core level photoexcitation for the spillover of the bulk Si electronic states to the SiO layer, (3) changes of band bending result from the excess minority carrier recombination at the oxidation-induced vacancy site when turning on and off the X-ray irradiation, and (4) a metastable chemisorbed O species (Pb1-paul) plays a decisive role in combining two kinds of the reaction loops of single- and double-step oxidation. Based on the experimental results, the unified Si oxidation reaction model mediated by point defect generation [Jpn. J. Appl. Phys. 59, SM0801 (2020)] is extended from a viewpoint of (a) the excess minority carrier recombination at the oxidation-induced vacancy site and (b) the trapping-mediated adsorption through the chemisorbed O species at the SiO/Si interface.
赤沢 第輔; 佐々木 岳彦*; 長坂 将成*; 志賀 基之
Journal of Chemical Physics, 156(4), p.044202_1 - 044202_7, 2022/01
被引用回数:8 パーセンタイル:62.23(Chemistry, Physical)セルロースの水和構造は、分子レベルでのバイオマスセルロースの加水分解メカニズムの理解のために非常に重要である。本論文では、セルロースの2糖の単量体であるセロビオースの水溶液について、そのX線吸収分光法(XAS)スペクトルの測定と理論計算を組み合わせた研究について報告する。測定に関する項目では高解像度の炭素K端のXASスペクトルの測定について報告する。理論計算の項目では、第一原理分子動力学計算と励起状態計算を用いてXASのシミュレーションを求める手法と結果ついて論ずる。セロビオースのXASには289.3eV, 290.7eV, 293.6eVの3つのピークがあり、それぞれアルコール基,ヘミアセタール基,アルコール及びヘミアセタール基の両方に対応することがわかった。さらに、25 Cから60Cまでの温度変化に対してスペクトルの高さが規則的に変化し、それがセロビオースと水分子の水素結合の数の変化を表していることがわかった。このスペクトルの変化は様々な環境におけるセルロースの水和構造を知るための情報として有用であると考えられる。
Cから60Cまでの温度変化に対してスペクトルの高さが規則的に変化し、それがセロビオースと水分子の水素結合の数の変化を表していることがわかった。このスペクトルの変化は様々な環境におけるセルロースの水和構造を知るための情報として有用であると考えられる。
長屋 勇輝*; 中津 裕貴*; 小倉 正平*; 島崎 紘太*; 植田 寛和; 武安 光太郎*; 福谷 克之
Journal of Chemical Physics, 155(19), p.194201_1 - 194201_6, 2021/11
被引用回数:1 パーセンタイル:4.26(Chemistry, Physical)We have developed a spin-polarized-hydrogen beam with a hexapole magnet. By combining the beam chopper and pulsed laser ionization detection, the time-of-flight of the hydrogen beam was measured, and the dependence of the beam profile on the velocity was acquired, which was consistent with the beam trajectory simulations. The spin polarization of the beam was analyzed by using the Stern-Gerlach-type magnet in combination with the spatial scan of the detection laser. The spin polarization was about 95% at a focusing condition due to the hexapole magnet. The polarization was, on the other hand, reduced to about 70% for the beam at higher velocities, which is consistent with simulation results.
 study of nuclear quantum effects on sub- and supercritical water
study of nuclear quantum effects on sub- and supercritical waterThomsen, B.; 志賀 基之
Journal of Chemical Physics, 155(19), p.194107_1 - 194107_11, 2021/11
被引用回数:8 パーセンタイル:47.53(Chemistry, Physical)The structures of water in the ambient, sub-, and supercritical conditions at various densities were studied systematically by path integral molecular dynamics simulations. It was found that the nuclear quantum effects (NQEs) have significant impact on the structure of hydrogen bonds in close contact, not only in the ambient condition but also in the sub-, and supercritical conditions. The NQEs on the structure beyond the hydrogen bond contact are important in ambient water, but not much for water in the sub-, and supercritical conditions. The NQEs are furthermore important for determining the number of hydrogen bonds in the ambient conditions, this role is however diminished in the sub- and supercritical conditions. The NQEs does nevertheless show their importance in determining the intramolecular structure of water and the close contact structures of the hydrogen bonds, even at sub- and supercritical conditions. Using the RPBE-D3 functional, the computed radial distribution functions for the ambient water are in excellent agreement with experimental data, upgrading our previous results using the BLYP-D2 functional [Machida, et al., J. Chem. Phys. 148, 102324 (2018)]. The computed radial distribution functions for water in the sub- and supercritical conditions were carefully compared with experiment. In particular, we found that the first peak in hydrogen pair distribution functions matches only when the NQEs are taken into account.
小林 恵太; 永井 佑紀; 板倉 充洋; 志賀 基之
Journal of Chemical Physics, 155(3), p.034106_1 - 034106_9, 2021/07
被引用回数:13 パーセンタイル:67.66(Chemistry, Physical)自己学習ハイブリッドモンテカルロ法は機械学習力場を利用することにより、第一原理計算の高速化を可能にする手法である。今回、自己学習ハイブリッドモンテカルロ法をNPTアンサンブルに適用し、液体シリカの解析を行った。本論文では自己学習ハイブリッドモンテカルロ法を用いることにより、厳密に第一原理計算の精度を保ちながら、高速に液体シリカの配置空間のサンプリングが可能であることを示した。また、液体シリカの構造因子の計算を行い実験データとの比較を行ったところ、両者はよい一致を見せた。
path integral molecular dynamicsThomsen, B.; 志賀 基之
Journal of Chemical Physics, 154(8), p.084117_1 - 084117_10, 2021/02
被引用回数:14 パーセンタイル:70.46(Chemistry, Physical)第一原理経路積分分子動力学シミュレーションにより、室温条件での水の同位体置換体の酸性度定数に対する核量子効果を計算した。この手法は、実験的に測定された液体DOの酸性度定数を再現するだけでなく、希少性のために不明な液体TOや、HDOおよびHTO水溶液の酸性度定数の理論的予測も可能にする。計算結果は、核量子効果が酸性度定数の絶対的評価において不可欠な役割を果たすことを示している。
野口 良史*; 樋山 みやび*; 志賀 基之; 秋山 英文*; 杉野 修*
Journal of Chemical Physics, 153(20), p.201103_1 - 201103_6, 2020/11
被引用回数:2 パーセンタイル:8.04(Chemistry, Physical)第一原理分子動力学シミュレーションを用いて、第一励起状態のオキシルシフェリン水溶液の3つの異性体の安定化メカニズムを調べた。フェノラート-ケト異性体のみが励起状態で水分子に引き付けられ、近くの水分子との水素結合の数を増やすことによって安定化された。励起状態で最も安定な異性体はフェノラート-ケトであり、フェノラート-エノールおよびフェノール-エノラート異性体は、フェノラート-ケトよりもエネルギーがそれぞれ0.38eVおよび0.57eV高かった。この結果は、フェノラート-エノールが最も安定な異性体である基底状態の場合とは対照的である。
赤浜 裕一*; 宮川 仁*; 谷口 尚*; 佐野 亜沙美; 町田 真一*; 服部 高典
Journal of Chemical Physics, 153(1), p.014704_1 - 014704_5, 2020/07
被引用回数:11 パーセンタイル:48.91(Chemistry, Physical)室温下で3.2GPaまでの圧力における黒リンの構造解析を粉末中性子回折によって行った。高圧下のリン原子の結合距離と結合角を精密に決め、これまでのX線単結晶解析の結果[Brown and Randqvist, Acta Cryst. 19, 684 (1965)]と一致することを確認した。結晶格子は異方的な圧縮を示すが、P1-P2およびP1-P3結合距離は圧力によらずほぼ一定であり、P3-P1-P2結合角のみ顕著に減少した。この結果から、Cartzら[J. Chem. Phys. 71, 1718 (1979)]により報告されている結合距離との顕著なずれは、解消された。我々の構造データは、高圧下における黒リンのDirac半金属状態を明らかにするに役立つ。
O山川 紘一郎; 那須 裕一*; 鈴木 菜摘*; 清水 元希*; 荒川 一郎*
Journal of Chemical Physics, 152(17), p.174310_1 - 174310_13, 2020/05
被引用回数:2 パーセンタイル:8.04(Chemistry, Physical)超高真空下テラヘルツ・赤外吸収分光装置を開発し、この装置を用いてAr固体中に分離したDOクラスターの吸収スペクトルを測定した。2, 3, 4量体によるテラヘルツ吸収ピークを帰属するため、スペクトルの温度依存性とDO希釈率依存性を分析した。帰属の妥当性は、ONIOM法を用いた第一原理計算を用いて検証した。これにより、2量体の全ての分子間振動モード、3量体と4量体の赤外吸収強度が大きい分子間振動モードを同定することができた。Ar固体中にDOクラスターを分離後、Arのみを昇華してDO氷を生成した。昇華温度と希釈率を変えてDO氷を生成し、テラヘルツスペクトルから結晶度を定量的に評価することで、結晶化にはDO単量体の拡散が決定的なプロセスであることを明らかにした。
 irradiation
irradiation岡崎 宏之*; 垣谷 健太*; 木全 哲也*; 出崎 亮*; 越川 博*; 松村 大樹; 山本 春也*; 八巻 徹也*
Journal of Chemical Physics, 152(12), p.124708_1 - 124708_5, 2020/03
被引用回数:5 パーセンタイル:24.42(Chemistry, Physical)X-ray absorption spectroscopy measurements were performed for the C K-edge of Pt nanoparticles on Ar-irradiated carbon supports in order to elucidate the origin of improved catalyst performance after the introduction of vacancies into the carbon support. We observed a change in the electronic structure at the interface between the Pt nanoparticles and the carbon support after vacancy introduction, which is in good agreement with theoretical results. The results indicated that vacancy introduction resulted in a drastic change in the Pt-C interactions, which likely affected the d-band center of the Pt nanoparticles and led to the enhancement of the oxygen reduction reaction in catalysts.
(101) surface長塚 直樹*; Wilde, M.*; 福谷 克之
Journal of Chemical Physics, 152(7), p.074708_1 - 074708_6, 2020/02
被引用回数:13 パーセンタイル:59.54(Chemistry, Physical)Hydrogenation of TiO enhances its visible photoabsorption, leading to efficient photocatalytic activity. However, the role of hydrogen has not been fully understood. The anatase TiO(101) surface treated by hydrogen ion irradiation at 500 eV was investigated by photoemission spectroscopy and nuclear reaction analysis. Hydrogen irradiation induces an in-gap state 1-1.6 eV below the Fermi level and a downward band bending of 0.27 eV. The H depth profile at 300 K shows a surface peak with an H amount of (2.9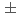 0.3)
0.3)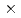 10
10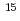 cm
cm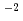 with little concentration in a deeper region. At 200 K, on the other hand, the H depth profile shows a maximum at about 1 nm below the surface corresponding to an H amount of (6.10.3)10 cm along with a broad distribution extending to 50 nm at an average concentration of 0.8 at. %. These results show that H diffusion in anatase TiO is much faster than in rutile TiO [Y. Ohashi, J. Phys. Chem. C 123, 10319-10324 (2019)]. The H diffusion coefficient at 200 K is determined to be 2.70.110
with little concentration in a deeper region. At 200 K, on the other hand, the H depth profile shows a maximum at about 1 nm below the surface corresponding to an H amount of (6.10.3)10 cm along with a broad distribution extending to 50 nm at an average concentration of 0.8 at. %. These results show that H diffusion in anatase TiO is much faster than in rutile TiO [Y. Ohashi, J. Phys. Chem. C 123, 10319-10324 (2019)]. The H diffusion coefficient at 200 K is determined to be 2.70.110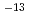 m
m s
s .
.
杉本 建*; 那須 裕一*; 荒川 一郎*; 山川 紘一郎
Journal of Chemical Physics, 150(18), p.184302_1 - 184302_5, 2019/05
被引用回数:4 パーセンタイル:17.43(Chemistry, Physical)メタンの結晶相IIの赤外吸収スペクトルを測定し、既報の回転振動ピークや振動-振動ピークに加えて、アニール後の急冷によって顕著なQ(2)ピークを検出することに成功した。R(0)、R(1)、Q(2)ピークの積分強度は2つの指数関数の線形結合の時間依存性を示した。これは、低エネルギー回転準位を占める3つの核スピン異性体間の相互変換を明らかに示している。
吉田 亨次*; 井上 拓也*; 鳥越 基克*; 山田 武*; 柴田 薫; 山口 敏男*
Journal of Chemical Physics, 149(12), p.124502_1 - 124502_10, 2018/09
被引用回数:4 パーセンタイル:14.97(Chemistry, Physical)異なる幾つかの、グリシン濃度, pH、および充填率(=グリシン溶液の質量/MCM-41の乾燥質量))をパラメーターとして、メソポーラスシリカ(MCM-41)に閉じ込められたグリシン水溶液の示差走査熱量測定、X線回折および準弾性中性子散乱(QENS)を305-180Kの温度範囲で実施して、グリシン水溶液の熱的挙動, 構造、および動的特性に対する閉じ込め効果を検討した。
