


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
天野 由記; Sachdeva, R.*; Gittins, D.*; Anantharaman, K.*; Lei, S.*; Valentin-Alvarado, L. E.*; Diamond, S.*; 別部 光里*; 岩月 輝希; 望月 陽人; et al.
Environmental Microbiome (Internet), 19, p.105_1 - 105_17, 2024/12
被引用回数:0 パーセンタイル:0.00(Genetics & Heredity)Underground research laboratories (URLs) provide a window on the deep biosphere and enable investigation of potential microbial impacts on nuclear waste, CO and H stored in the subsurface. We carried out the first multi-year study of groundwater microbiomes sampled from defined intervals between 140 and 400 m below the surface of the Horonobe and Mizunami URLs, Japan. The Horonobe and Mizunami microbiomes are dissimilar, likely because the Mizunami URL is hosted in granitic rock and the Horonobe URL in sedimentary rock. Despite this, hydrogen metabolism, rubisco-based CO fixation, reduction of nitrogen compounds and sulfate reduction are well represented functions in microbiomes from both URLs, although methane metabolism is more prevalent at the organic- and CO-rich Horonobe URL. We detected near-identical genotypes for approximately one third of all genomically defined organisms at multiple depths within the Horonobe URL. This cannot be explained by inactivity, as in situ growth was detected for some bacteria, albeit at slow rates. Given the current low hydraulic conductivity and groundwater compositional heterogeneity, ongoing inter-site strain dispersal seems unlikely. Alternatively, the Horonobe URL microbiome homogeneity may be explained by higher groundwater mobility during the last glacial period. Genotypically-defined species closely related to those detected in the URLs were identified in three other subsurface environments in the USA. Thus, dispersal rates between widely separated underground sites may be fast enough relative to mutation rates to have precluded substantial divergence in species composition. Species overlaps between subsurface locations on different continents constrain expectations regarding the scale of global subsurface biodiversity. Overall, microbiome and geochemical stability over the study period has important implications for underground storage applications.
and H stored in the subsurface. We carried out the first multi-year study of groundwater microbiomes sampled from defined intervals between 140 and 400 m below the surface of the Horonobe and Mizunami URLs, Japan. The Horonobe and Mizunami microbiomes are dissimilar, likely because the Mizunami URL is hosted in granitic rock and the Horonobe URL in sedimentary rock. Despite this, hydrogen metabolism, rubisco-based CO fixation, reduction of nitrogen compounds and sulfate reduction are well represented functions in microbiomes from both URLs, although methane metabolism is more prevalent at the organic- and CO-rich Horonobe URL. We detected near-identical genotypes for approximately one third of all genomically defined organisms at multiple depths within the Horonobe URL. This cannot be explained by inactivity, as in situ growth was detected for some bacteria, albeit at slow rates. Given the current low hydraulic conductivity and groundwater compositional heterogeneity, ongoing inter-site strain dispersal seems unlikely. Alternatively, the Horonobe URL microbiome homogeneity may be explained by higher groundwater mobility during the last glacial period. Genotypically-defined species closely related to those detected in the URLs were identified in three other subsurface environments in the USA. Thus, dispersal rates between widely separated underground sites may be fast enough relative to mutation rates to have precluded substantial divergence in species composition. Species overlaps between subsurface locations on different continents constrain expectations regarding the scale of global subsurface biodiversity. Overall, microbiome and geochemical stability over the study period has important implications for underground storage applications.
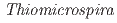 sp. strain V2501 isolated from 250 m below the ground level in Horonobe, Hokkaido, Japan
sp. strain V2501 isolated from 250 m below the ground level in Horonobe, Hokkaido, Japan上野 晃生*; 佐藤 聖*; 玉村 修司*; 村上 拓馬*; 猪股 英紀*; 玉澤 聡*; 天野 由記; 宮川 和也; 長沼 毅*; 五十嵐 敏文*
Microbiology Resource Announcements (Internet), 13(11), p.e00108-24_1 - e00108-24_4, 2024/11
A thiosulfate oxidizing bacterium, sp. strain V2501, was isolated from groundwater collected in a terrestrial deep subsurface environment. This strain grows chemolithoautotrophically in media containing NaHCO as its sole carbon source. Here, we report a 2,240,851 bp complete genome sequence of strain V2501.
as its sole carbon source. Here, we report a 2,240,851 bp complete genome sequence of strain V2501.
Jaffe, A. L.*; Thomas, A. D.*; He, C.*; Keren, R.*; Valentin-Alvarado, L. E.*; Munk, P.*; Bouma-Gregson, K.*; Farag, I. F.*; 天野 由記; Sachdeva, R.*; et al.
mBio, 12(4), p.e00521-21_1 - e00521-21_21, 2021/08
被引用回数:28 パーセンタイル:87.18(Microbiology)Candidate Phyla Radiation (CPR) bacteria are small, likely episymbiotic organisms found across Earth's ecosystems. Despite their prevalence, the distribution of CPR lineages across habitats and the genomic signatures of transitions amongst these habitats remain unclear. Hear, we expand the genome inventory for Absconditabacteria (SR1), Gracilibacteria, and Saccharibacteria (TM7), CPR bacteria known to occur in both animal-associated and environmental microbiomes, and investigate variation in gene content with habitat of origin. By overlaying phylogeny with habitat information, we show that bacteria from these three lineages have undergone multiple transitions from environmental habitats into animal microbiomes. Based on co-occurrence analyses of hundreds of metagenomes, we extend the prior suggestion that certain TM7 have broad bacterial host ranges and constrain possible host relationships for SR1 and Gracilibacteria. Full-proteome analyses show that animal-associated TM7 have smaller gene repertoires than their environmental counterparts and are enriched in numerous protein families, including those likely functioning in amino acid metabolism, phage defense, and detoxification of peroxide. In contrast, some freshwater TM7 encodea putative rhodopsin. For protein families exhibiting the clearest patterns of differential habitat distribution, we compared protein and species phylogenies to estimate the incidence of lateral gene transfer and genomic loss occurring over the species tree. These analyses suggest that habitat transitions were likely not accompanied by large transfer or loss events, but rather were associated with continuous proteome remodeling. Thus, we speculate that CPR habitat transitions were driven largely by availability of suitable host taxa, and were reinforced by acquisition and loss of some capacities.
Al-Shayeb, B.*; Sachdeva, R.*; Chen, L.-X.*; Ward, F.*; Munk, P.*; Devoto, A.*; Castelle, C. J.*; Olm, M. R.*; Bouma-Gregson, K.*; 天野 由記; et al.
Nature, 578(7795), p.425 - 431, 2020/02
被引用回数:295 パーセンタイル:99.46(Multidisciplinary Sciences)Phage typically have small genomes and depend on their bacterial hosts for replication. We generated metagenomic datasets from many diverse ecosystems and reconstructed hundreds of huge phage genomes, between 200 kbp and 716 kbp in length. Thirty four genomes were manually curated to completion, including the largest phage genomes yet reported. Expanded genetic repertoires include diverse and new CRISPR-Cas systems, tRNAs, tRNA synthetases, tRNA modification enzymes, initiation and elongation factors and ribosomal proteins. Phage CRISPR have the capacity to silence host transcription factors and translational genes, potentially as part of a larger interaction network that intercepts translation to redirect biosynthesis to phage-encoded functions. Some phage repurpose bacterial systems for phage-defense to eliminate competing phage. We phylogenetically define seven major clades of huge phage from human and other animal microbiomes, oceans, lakes, sediments, soils and the built environment. We conclude that large gene inventories reflect a conserved biological strategy, observed across a broad bacterial host range and resulting in the distribution of huge phage across Earth's ecosystems.
and metal transformations associated with novel bacteria and archaea in deep terrestrial subsurface sedimentsHernsdorf, A. W.*; 天野 由記; 宮川 和也; 伊勢 孝太郎; 鈴木 庸平*; Anantharaman, K.*; Probst, A. J.*; Burstein, D.*; Thomas, B. C.*; Banfield, J. F.*
ISME Journal, 11, p.1915 - 1929, 2017/03
被引用回数:107 パーセンタイル:96.15(Ecology)地層処分システムにおける微生物影響の可能性を評価するために、北海道の幌延深地層研究センター地下施設を利用して、堆積岩地下の生態系における微生物群集構造と代謝機能について調査を行った。全体として、微生物生態系は多様な系統群からなる微生物種で構成されており、その多くはこれまで培養されていない生物門に属していることが示された。大部分の微生物種は、酸化型[NiFe]ヒドロゲナーゼあるいはフェレドキシンをベースとする代謝経路を可能にする電子分岐型[FeFe]ヒドロゲナーゼを介して水素代謝をおこなうことが明らかになった。水素代謝と関連して、多くの微生物が炭素,窒素,鉄および硫黄を代謝することが推定された。特に、ANME-2dというメタン酸化を行う古細菌として知られている未培養微生物が、鉄関連の代謝反応を行う可能性が示唆された。得られた結果から、幌延堆積岩環境における微生物群集の生態学的概念モデルを推定した。
増渕 隆*; 渡口 和樹*; 林 秀謙*; 池永 裕*; 佐藤 勝也; 大野 豊
JAEA-Review 2015-022, JAEA Takasaki Annual Report 2014, P. 106, 2016/02
We conducted the gene function analysis of the high ethyl caproate producing sake yeast mutant (No.227) generated by ion beam breeding in order to develop a new method for sake yeast screening and find factors contributing the reduced ability to alcohol fermentation. The genome sequences of the No.227 and Kyokai 901 (its parental strain) were determined by a whole-genome shotgun strategy using pyrosequencing method. And the determined sequences were compared with that of the sake yeast strain Kyokai 7, which is characterized by the fermentation properties. Consequently, the No.227 carried mutations in the 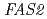 and
and 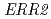 genes as heterozygous state. It was suggested that these mutations in the and genes in the No.227 might be attributed to the high ethyl caproate production and the low alcohol fermentation ability, respectively.
genes as heterozygous state. It was suggested that these mutations in the and genes in the No.227 might be attributed to the high ethyl caproate production and the low alcohol fermentation ability, respectively.
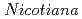 species with different ploidy levels revealed by 5S rDNA spacer sequences and FISH/GISH
species with different ploidy levels revealed by 5S rDNA spacer sequences and FISH/GISH北村 智; 田中 淳; 井上 雅好*
Genes and Genetic Systems, 80(4), p.251 - 260, 2005/08
被引用回数:10 パーセンタイル:19.66(Biochemistry & Molecular Biology)タバコ属植物における倍数化の経緯を探るため、4倍体野生タバコ種において、高等生物に必須の遺伝子である5S rDNAのスペーサー領域を単離しシーケンス解析した。既に解析済みであった2倍体タバコ種におけるスペーサー配列と比較することにより、2倍体種と4倍体種からなる複数のグループが形成された。また5S rDNAの座乗染色体を調べたところ、グループ内の種は類似した染色体に5S rDNAが位置することがわかった。これらの結果から、同一グループに分類された倍数性の異なる種は、5S rDNAを基準にすると、非常に系統学的に近い関係にあると言える。この結果が、5S rDNAという一つの遺伝子だけでなくゲノム全体に関しても言えるかどうかを調査するために、GISHによるゲノムレベルの解析を行った。その結果、シーケンス解析で認められたグループは、ゲノムレベルでも非常に近縁関係にあることがわかった。
木村 英雄; 酒井 智*
JAERI-Data/Code 2004-001, 32 Pages, 2004/03
 JAERI-Data-Code-2004-001.pdf:1.34MB
JAERI-Data-Code-2004-001.pdf:1.34MB
現在、生物学の研究は、ゲノムと呼ばれる遺伝子のセットを用いて生命全体の働きを明らかにしていく段階に入っている。数千に及ぶ遺伝子がもたらす生命の全体像を明らかにするには、遺伝子の相互作用から生み出される莫大な情報を解析しなくてはならない。今までの実験による解析方法では、この量の情報解析を現実的な時間で行うのは不可能である。そこで注目されているのが、コンピュータによる解析である。バイオインフォマティクスと呼ばれる分野は、生物学と情報学とが融合した新しい分野であり、今急速に発展している。本書では、バイオインフォマティクス分野の支援としてITBL利用推進室が行っている、データベースの整備、及びWeb経由でアプリケーションを実行するためのシステム(アプリケーション実行Webシステム)について報告する。
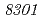 in the radioresistant bacterium
in the radioresistant bacterium 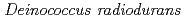
Islam, M. S.*; Hua, Y.*; 大庭 寛史; 佐藤 勝也; 菊地 正博; 柳沢 忠*; 鳴海 一成
Genes and Genetic Systems, 78(5), p.319 - 327, 2003/10
被引用回数:16 パーセンタイル:30.52(Biochemistry & Molecular Biology)放射線抵抗性細菌デイノコッカス・ラジオデュランスKD8301株から単離された挿入配列ISを解析した。ISは、IS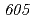 /IS
/IS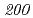 グループに属する新規挿入配列であり、長さが1,736塩基対であった。Inverse PCR法での解析により、ISの挿入標的部位は"TTGAT"であることがわかった。ISラジオデュランスの野生株R
グループに属する新規挿入配列であり、長さが1,736塩基対であった。Inverse PCR法での解析により、ISの挿入標的部位は"TTGAT"であることがわかった。ISラジオデュランスの野生株R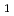 , MR, KRでのISのゲノム内分布を調べたところ、R株の公開ゲノム配列情報と矛盾しており、今回調べたR株ではISが1コピーしか存在しなかった。ISは挿入部位に存在する遺伝子を破壊しており、ゲノムに変異を起こす原因となりうるが、大規模のゲノム再構成を引き起こしてはいなかった。
, MR, KRでのISのゲノム内分布を調べたところ、R株の公開ゲノム配列情報と矛盾しており、今回調べたR株ではISが1コピーしか存在しなかった。ISは挿入部位に存在する遺伝子を破壊しており、ゲノムに変異を起こす原因となりうるが、大規模のゲノム再構成を引き起こしてはいなかった。
Battista, J. R.*; Cox, M. M.*; Daly, M. J.*; 鳴海 一成; Radman, M.*; Sommer, S.*
Science, 302(24), p.567 - 568, 2003/10
デイノコッカス・ラジオデュランスの細胞内核様体がドーナツ状構造をとり、この構造が放射線抵抗性とかかわりがあるとする論文がScienceの1月号に掲載された。しかし、この論文の結論は電子顕微鏡による切片の観察のみをもとに構築されたものであり、他の実験的証拠に乏しく、しかも未だ同定されていない「正確なDNA末端再結合によるDNA2本鎖修復」に関する彼らの仮説は、過去の実験的データを誤解して解釈したものを拠り所にしている。細胞中のゲノムコピー数に関しても、過去の実験データを無視して議論しており、彼らの仮説と研究結果は受け入れ難い。細胞内DNAの構造とDNA修復との関係を調べることは重要であるという認識は一致するところであり、より実りの多い遺伝学や分子生物学的実験と相補しながら、研究をさらに進めることが必要である。
鳴海 一成
Trends in Microbiology, 11(9), p.422 - 425, 2003/09
被引用回数:53 パーセンタイル:90.46(Biochemistry & Molecular Biology)放射線抵抗性細菌デイノコッカス・ラジオデュランスのDNAマイクロアレイによるトランスクリプトーム解析と、電子顕微鏡観察による核様体の特異的形態変化についての発見が、最近トップジャーナルで相次いで報告された。これらの研究は、デイノコッカス・ラジオデュランスの放射線耐性機構の解明に進展をもたらしたが、この細菌がなぜ放射線に強いのかについて、より詳細な実験的証拠をもとにした説明が必要なのであろうか?実りの多い遺伝学的並びに生化学的アプローチによるさらなる研究が、放射線抵抗性細菌のDNA修復機構についてのより深い理解のために必要である。
由良 敬
プロテオミクスの最新技術, p.93 - 101, 2002/11
ゲノム配列の決定は生物学に大きな影響をもたらす。ゲノム配列とはそのゲノムをもつ生物種の設計図であり、ゲノム配列を解読することで、その生物がどのようにして構成されているのかが理解できるからである。1995年にHaemophilus influenzaeの全ゲノムがはじめて決定された時点で、生物学者が直面した問題は、ゲノム配列がわかっても、残念ながらその中に何が書かれているのかがわからないということであった。それまでの遺伝学及び生化学の知見では、理解できない情報が厖大に存在する。これらの情報を従来と同じ遺伝学及び生化学の手法のみで解析していくのは不可能に近い。そこで脚光を浴びるようになったのが生命情報学である。本章では生命情報学の現状と未来を解説する。
鳴海 一成; 菊地 正博; 舟山 知夫; 佐藤 勝也
放射線生物研究, 34(4), p.401 - 418, 1999/00
生物は常にDNA損傷を被る環境に曝されており、生命の維持はDNA損傷をどれだけ効率よく正確に修復できるかに懸かっている。したがって、生物のDNA修復能力の限界を見極めることは、巧妙な生命現象へのわれわれのより深い理解にとって大切なことであるのみならず、生命維持機能を増強する方法の開発に方向性を与えることにも繋がる。今までともすれば奇異な目で見られることもあった放射線抵抗性細菌デイノコッカス・ラジオデュランスを、ほかの生物と同様に放射線生物研究の対象として扱い、ほかの生物とどこが同じでどこが違うのかを明らかにして、その極めて高いDNA修復能力を分子レベルで解明する研究が、最近になって急速に進展しつつある。本稿では、デイノコッカス・ラジオデュランスの特徴的側面、及びわれわれの研究室で得られた最新のデータをまじえ、DNA修復遺伝子群の解析に関する最近の進展状況について述べる。
宮本 旬子*; 廣瀬 玉紀; 秋山 征夫*; 福井 希一*
Proceedings of 16th International Botanical Congress, P. 618, 1999/00
ゲノム画像情報には、塩基配列情報のみならず、タンパク質とDNAの複合体である染色体、さらには細胞核に至るまでの高次元の構造情報も含まれる。そこで染色体画像のデータベースシステム「CAJ」を構築した。ゲノム解析研究、生物多様性などバイオテクノロジーの著しい進歩により一般社会からも遺伝子の働きと遺伝情報の総体であるゲノム(細胞核・染色体)が注目されている。しかしながら当該分野の研究者においてすらゲノム(細胞核)や染色体の実際の姿を知る機会は極めて限られており、大部分の情報は専門の研究者個々人のファイル中に個別的・分散的に保存され、一般には触れにくい状況にある。本データベースは、ゲノム関連研究者はもちろん、種々の生物科学の進展に関連して必要とされるゲノム情報を多分野にわたる人々にインターネットを通じて提供し、今後各種トピックのコンテンツを順次追加し、そのニーズに応えるものである。
天野 由記; Sachdeva, R.*; Gittins, D.*; Anantharaman, K.*; Lei, S.*; 別部 光里*; 望月 陽人; Thomas, B. C.*; 鈴木 庸平*; Banfield, J. F.*
no journal, ,
Underground research laboratories (URLs) provide a window on the deep biosphere and enable investigation of potential microbial impacts on geological disposal, CO and H stored in the subsurface. We carried out the first multi-year study of groundwater microbiomes sampled from defined intervals between 140 and 400 m below the surface of the Horonobe and Mizunami URLs, Japan. We reconstructed draft genomes for 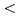 90% of all organisms detected over a four-year period. The Horonobe and Mizunami microbiomes are dissimilar, likely because the Mizunami URL is hosted in granitic rock and the Horonobe URL in sedimentary rock. Despite this, hydrogen metabolism, rubisco-based CO fixation, reduction of nitrogen compounds and sulfate reduction are well represented functions in microbiomes from both URLs, although methane metabolism is more prevalent at the organic- and CO -rich Horonobe URL. High fluid flow zones and proximity to subsurface tunnels apparently impact microbial community composition. We detected near-identical genotypes for approximately one third of genomically defined organisms at multiple depths within the Horonobe URL. Sequencing data indicate that at least some of the bacteria are growing, albeit slowly. Genotypically-defined species closely related to those detected in the URLs were identified in three other subsurface environments in the USA. Thus, dispersal rates may be fast enough relative to mutation rates to have limited substantial species divergence. Hydraulic and isotopic measurements predict inter-site movement of groundwater over thousands of years, but it is uncertain whether the permeability is sufficient for transport of microbial cells. Alternatively, Horonobe URL microbiome homogeneity may be explained by dispersal during last glacial period.
90% of all organisms detected over a four-year period. The Horonobe and Mizunami microbiomes are dissimilar, likely because the Mizunami URL is hosted in granitic rock and the Horonobe URL in sedimentary rock. Despite this, hydrogen metabolism, rubisco-based CO fixation, reduction of nitrogen compounds and sulfate reduction are well represented functions in microbiomes from both URLs, although methane metabolism is more prevalent at the organic- and CO -rich Horonobe URL. High fluid flow zones and proximity to subsurface tunnels apparently impact microbial community composition. We detected near-identical genotypes for approximately one third of genomically defined organisms at multiple depths within the Horonobe URL. Sequencing data indicate that at least some of the bacteria are growing, albeit slowly. Genotypically-defined species closely related to those detected in the URLs were identified in three other subsurface environments in the USA. Thus, dispersal rates may be fast enough relative to mutation rates to have limited substantial species divergence. Hydraulic and isotopic measurements predict inter-site movement of groundwater over thousands of years, but it is uncertain whether the permeability is sufficient for transport of microbial cells. Alternatively, Horonobe URL microbiome homogeneity may be explained by dispersal during last glacial period.
横谷 明徳
no journal, ,
放射線を照射された生体中には、ゲノム中の遺伝子分子(DNA)に化学的な構造変化(損傷)が高い頻度で生じる。一方、これらのDNA損傷を効率よく酵素的に除去し元に戻す仕組み(修復機構)を細胞は備えている。しかし修復がうまく働かない場合には、突然変異など重大な影響が現れる。フクシマにおける低線量放射線影響がここまで大きな問題になっている理由のひとつは、DNA損傷とその後の生体修復について未だ解明が遅れていることに起因する。われわれはこれまで、軟X線をプローブとして利用しDNA損傷生成の初期過程の解明を目指した研究を行ってきた。特に元素選択的なイオン化により生起するDNA損傷がどのような違いをもたらすのかについて、電子常磁性共鳴法や質量分析法による生成物分析から追跡するとともに、実際の生体修復酵素と照射DNAの反応を調べている。本講演では、これらの研究成果を通して得た知見を元に、内殻イオン化後の多価イオン原子及び後続のAuger過程により生じた低速電子がDNA損傷をどのように誘発するかについて考察するとともに、ゲノムDNA分子上の数nm程度に複数の損傷が分布する"クラスターDNA損傷"と難修復性との関連について述べる。
横谷 明徳; 鈴木 啓司*
no journal, ,
細胞のガン化に深く関係するとされるゲノム不安定性は、放射線照射後にこれらのDSBが修復されたにも関わらず、細胞分裂が繰り返された後の子孫細胞にも表れる現象である。DNA損傷が修復され細胞分裂が繰り返された後に、どのようなメカニズムで細胞は放射線が照射された記憶を保持できるのか、その詳細はほとんど明らかになっていない。本研究では、ゲノム中に生じたDSBが、その修復過程で、大規模なゲノム再構成を引き起こした場合、その近傍には、ヒストンのリン酸化などの化学修飾が何らかのエピジェネティックなメモリーとして細胞分裂後にも依然として残っているのではないか?ということを作業仮説とした。この仮説を検証するため、放射線照射によりHPRT遺伝子座を含む大規模な欠失が生じたヒトの突然変異細胞を試料として用い、最初の作業として欠失部位をRT-PCR法により精密に特定することを試みた。16種類のPCRプローブを用いて欠失部位の特定を試みた結果、特定の、ゲノム欠失部位については250kbpまで絞り込むことに成功した。さらに、これらの欠失部位の精密な位置情報に基づき、今後展開するChip Assay法によるヒストンの化学修飾状態の空間的な広がりの探索法を紹介する。
佐藤 勝也; 武田 喜代子*; 鳴海 一成*; 大津 直子*; 横山 正*
no journal, ,
根粒菌はマメ科植物と共生し、窒素栄養をマメ科植物に供給する。現在、化学肥料からの脱却を目指し、東南アジア等では、この共生関係を生かした根粒菌バイオ肥料の利用が奨励されている。しかし、化学肥料とは異なり、バイオ肥料は、輸送や保存時の高温による接種菌の活性低下等の問題があり改良が望まれている。これまで我々は、ダイズ根粒菌の温帯優良接種菌株である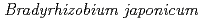 USDA110株を対象としたイオンビーム突然変異育種を行ってきた。そして、42
USDA110株を対象としたイオンビーム突然変異育種を行ってきた。そして、42 Cの液体培地中で少なくとも1週間は高い生存率を維持することが可能な高温耐性変異株の取得に成功した。本研究では、次世代シークエンサーを用いて高温耐性変異株の全ゲノムを解読し、野生株との塩基配列の比較によってDNA変異部位を同定した。ゲノムDNA塩基配列の比較解析の結果、高温耐性変異株M14において、1.27Mbpの逆位という大規模なゲノム構造変異、及び一塩基置換・欠失及び挿入などの18ヶ所の小規模なDNA塩基変異が存在することを明らかにした。これらの大規模な構造変異あるいは小規模な遺伝子変異がM14株の高温耐性に関与している可能性が考えられた。
Cの液体培地中で少なくとも1週間は高い生存率を維持することが可能な高温耐性変異株の取得に成功した。本研究では、次世代シークエンサーを用いて高温耐性変異株の全ゲノムを解読し、野生株との塩基配列の比較によってDNA変異部位を同定した。ゲノムDNA塩基配列の比較解析の結果、高温耐性変異株M14において、1.27Mbpの逆位という大規模なゲノム構造変異、及び一塩基置換・欠失及び挿入などの18ヶ所の小規模なDNA塩基変異が存在することを明らかにした。これらの大規模な構造変異あるいは小規模な遺伝子変異がM14株の高温耐性に関与している可能性が考えられた。
増淵 隆*; 日向 弘和*; 池永 裕*; 林 秀謙*; 佐藤 勝也; 大野 豊
no journal, ,
群馬県では、オリジナルの吟醸用清酒酵母を開発するために、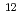 C
C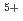 (220MeV)のイオンビーム照射による突然変異で新たな吟醸用清酒酵母の開発を行っており、これまでに吟醸酒特有の香気成分であるカプロン酸エチルを高生産する優良清酒酵母(No.227)を選抜した。しかし、カプロン酸エチル高生産酵母は発酵能が弱くなることがあり、また、変異株の選抜・機能評価は多大な手数と時間を必要とし、研究の遂行を困難にする一因となっている。本研究はイオンビーム変異により得られた酵母の全ゲノムDNA塩基配列を解読し、発酵特性等の機能とゲノム情報が既に明らかとなっている酵母と比較することで、酵母選抜のメルクマールとなる香気生成能・発酵能に関与する遺伝子群を特定すること目的として行った。
(220MeV)のイオンビーム照射による突然変異で新たな吟醸用清酒酵母の開発を行っており、これまでに吟醸酒特有の香気成分であるカプロン酸エチルを高生産する優良清酒酵母(No.227)を選抜した。しかし、カプロン酸エチル高生産酵母は発酵能が弱くなることがあり、また、変異株の選抜・機能評価は多大な手数と時間を必要とし、研究の遂行を困難にする一因となっている。本研究はイオンビーム変異により得られた酵母の全ゲノムDNA塩基配列を解読し、発酵特性等の機能とゲノム情報が既に明らかとなっている酵母と比較することで、酵母選抜のメルクマールとなる香気生成能・発酵能に関与する遺伝子群を特定すること目的として行った。
天野 由記; Diamond, S.*; Lavy, A.*; Anantharaman, K.*; 宮川 和也; 岩月 輝希; 別部 光里*; 鈴木 庸平*; Thomas, B. C.*; Banfield, J. F.*
no journal, ,
We investigated the microbiology two Japanese subsurface research sites and compared the major groups of organisms lacking cultivated representatives found from other subsurface sites, including a Colorado aquifer and deep aquifers underlying the Crystal Geyser. We analyzed metagenomic data 19 samples from the Horonobe site and 7 from the Mizunami site. DNA sequences from each sample were assembled independently and scaffolds encoding the ribosomal protein S3 sequence were identified. The major characteristic of the microbiology of the Mizumani site that distinguished it from the Horonobe site is local very high abundances of Nitrospirae, Parcubacteria, Ignavibacteria, ANME-2D and Micrarchaeota. In contrast, the Horonobe site has locations that are highly enriched in Altarchiales, Syntrophobacteriales, Atribacteria, ANME-2D and Methanogens. Beyond reshaping the Tree of Life, the societal importance of these discoveries remains little known. However, given the huge inventory of new groups of proteins and pathways in the genomes of these organisms, it is reasonable to anticipate major discoveries will hold relevance, for example, in terms of pharmaceutical discovery. Given the importance of the subsurface as a potential host environment for storage of nuclear waste, finding some commonality would indicate the general relevance of information from one site for prediction of the characteristics of other sites.
