


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
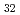 (Al, Zn)
(Al, Zn)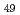 Approximant crystal; Influence of chemical disorder
Approximant crystal; Influence of chemical disorder清水 一行*; 山口 正剛; 赤丸 悟士*; 西村 克彦*; 阿部 李音*; 佐々木 泰祐*; Wang, Y.*; 戸田 裕之*
Scripta Materialia, 265, p.116730_1 - 116730_7, 2025/08
被引用回数:0 パーセンタイル:0.00(Nanoscience & Nanotechnology)The approximant crystal Mg(Al, Zn) (T-phase in Al-Zn-Mg alloys) holds the potential for enhancing both strength and hydrogen embrittlement resistance in aluminum alloys when present as nano-precipitates. Our previous computational exploration indicated strong hydrogen trapping but neglected the inherent chemical disorder of this approximant crystal. This study revisits hydrogen trapping in Mg(Al, Zn), directly including chemical disorder via special quasirandom structures. Density functional theory calculations reveal that, while chemical disorder introduces variations in trapping energies, the overall trend of strong trapping persists. Tetrahedral sites coordinated by Mg atoms exhibit particularly strong trapping, with smaller tetrahedral volumes correlating with stronger trapping due to enhanced Mg-H interactions. Multiple hydrogen occupation of these sites is also calculated, resulting in high hydrogen densities. Experimental validation using thermal desorption spectroscopy on a bulk Mg(Al, Zn) sample confirms hydrogen trapping, reinforcing the potential of this phase for designing advanced, hydrogen-resistant aluminum alloys.
海老原 健一; 藤原 比呂*; 清水 一行*; 山口 正剛; 戸田 裕之*
International Journal of Hydrogen Energy, 136, p.751 - 756, 2025/06
被引用回数:0高強度のAl-Zn-Mg合金へのSn添加は水素脆化を抑制する効果があるとの実験的報告があり、これは、Snの第二相粒子による水素吸収に起因すると考えられる。この事実を検証するため、第一原理計算で評価した水素の固溶エネルギーを取り入れた反応拡散方程式に基づくモデルを用い、アルミ中のSn相への水素侵入のシミュレーションを実施した。その結果、Sn第二相粒子による水素吸収が水素脆化を抑制するには、第二相粒子の水素固溶サイト濃度がAl相の5倍以上必要であることが分かった。このことから、実際にはSn第二相粒子のHE抑制効果は限定的であり、実験における水素脆化抑制には他の要因の影響が考えられる。
Shi, W.*; 町田 昌彦; 山田 進; 岡本 孝司
Progress in Nuclear Energy, 184, p.105710_1 - 105710_10, 2025/06
被引用回数:0 パーセンタイル:0.00(Nuclear Science & Technology)最近、原子炉建屋内の少ない観測点で測定された空間線量値から放射線源分布を逆推定する方法としてLASSO(Least Absolute Shrinkage and Selection Operator)が提案された。しかし、空間線量値を測定する際には誤差が含まれるが、この誤差が逆推定結果の精度にどのように影響するかは解析されていなかった。そこで本論文では、LASSOに対する不確実性解析を行い、Candesの理論に基づく不確実性推定関数を提案した。実際に、モンテカルロ法の1つであるParticle and Heavy Ion Transport code System (PHITS)を使用して得られた不確実性を持つ数値を入力値として用いたテスト計算を実施し、計算結果の誤差が提案された推定関数に従うことを示した。これにより、LASSOは推定された不確実性の範囲内で放射線源分布を求めることができる方法であることが確認できた。
 path-integral simulations of hydrogen-isotope diffusion in face-centred cubic metals
path-integral simulations of hydrogen-isotope diffusion in face-centred cubic metals君塚 肇*; 尾方 成信*; Thomsen, B.; 志賀 基之
Journal of Physics; Condensed Matter, 37(19), p.193001_1 - 193001_16, 2025/04
被引用回数:0 パーセンタイル:0.00(Physics, Condensed Matter)第一原理経路積分法を用いて、面心立方格子金属の水素同位体拡散係数を計算したところ、拡散係数の温度依存性が特異な形状をとることがわかった。これは温度依存性の異なる核量子効果の競合に起因しており、通常の同位体効果と逆同位体効果の間の異常なクロスオーバーのメカニズムを説明するものである。また、密度汎関数理論計算とほぼ同じ精度で、機械学習ベースの原子間ポテンシャルを用いて、近似的な量子ダイナミクスを記述する経路積分シミュレーションの結果も紹介する。この計算法は、様々な固体物質における水素同位体の量子的振る舞いを解明する道を開くものである。
高久 雄飛; 掛札 豊和*; 矢城 重夫; 木村 英雄; 久野 哲也
JAEA-Testing 2024-006, 31 Pages, 2025/03
 JAEA-Testing-2024-006.pdf:2.5MB
JAEA-Testing-2024-006.pdf:2.5MB
国立研究開発法人日本原子力研究開発機構(原子力機構)では、各部署に多種多様なサーバが存在しており、現場のサーバ管理者がセキュリティ対策をはじめとするサーバ管理業務を担っているため、業務負担や現場での運用コスト負担は無視できないものとなっている。また近年、国内外で多発しているセキュリティインシデントへの対策は、多くの機微情報を扱う原子力機構の業務を遂行する上で必要不可欠であるが、各サーバ管理者が能動的にこれらの措置を講ずることは困難なのが現状である。これらの課題を解決するため、コスト削減に観点を置いて機微情報を含まない一般情報システムの外部クラウド環境への移行を実施した。移行の結果、コストを大幅に削減しつつ円滑に外部クラウド環境へ移行するための条件を明らかにし、かつセキュリティインシデントへの対策を実現する基盤を整えた。
Yoshida, M.*; McDermott, R. M.*; Angioni, C.*; Camenen, Y.*; Citrin, J.*; Jakubowski, M.*; Hughes, J. W.*; 井戸村 泰宏; Mantica, P.*; Mariani, A.*; et al.
Nuclear Fusion, 65(3), p.033001_1 - 033001_132, 2025/02
被引用回数:4 パーセンタイル:95.90(Physics, Fluids & Plasmas)ITER Physics Basis出版以降のITPA輸送・閉じ込め(TC)グループにおけるプラズマ輸送と閉じ込めの物理理解と理論モデル開発の進展を、ITERと燃焼プラズマの予測・制御への貢献に焦点を当ててまとめた。本論文は、過去15年間のITPA TC共同実験/共同活動によって主に導かれた進歩について、一般的かつ合理的な概観を提供するものである。本論文は、ITPA TCグループの科学的な戦略とスコープ、主要な進歩の全体像から始まり、粒子輸送、不純物輸送、イオン・電子乱流熱輸送、運動量輸送、3次元磁場が輸送に与える影響、閉じ込めモード遷移、大域的閉じ込め、簡約化輸送モデルといった各研究分野の進歩を示す。
山口 瑛子; 奥村 雅彦; 河村 直己*; 高橋 嘉夫*
Science of the Total Environment, 964, p.178585_1 - 178585_13, 2025/02
粘土鉱物の吸着反応にはまだ未解明な点が多く残されており、その原因の一つは吸着サイトが複数存在することである。それぞれの吸着サイトの寄与量は吸着イオンの濃度に依存することが知られており、主に高濃度試料を分析する原子レベルでの研究結果と低濃度試料が多い環境試料の分析結果の包括的な理解に課題がある。そこで本研究では、放射光を用いた実験や第一原理計算を組み合わせることで、吸着濃度に応じて吸着サイト及び吸着イオンの局所構造が系統的に変化する様子を原子レベルで捉え、吸着イオンと粘土鉱物の相互作用がイオン結合的であることを示した。
システム計算科学センター 高性能計算技術利用推進室
JAEA-Review 2024-044, 121 Pages, 2025/01
JAEA-Review-2024-044.pdf:7.42MB
日本原子力研究開発機構では、原子力の総合的研究開発機関として原子力に係わるさまざまな分野の研究開発を行っており、これらの研究開発の多くにおいて計算科学技術が活用されている。日本原子力研究開発機構における計算科学技術を活用した研究開発の論文発表は、過去十数年にわたり、毎年度、全体の約2割を占めている。大型計算機システムはこの計算科学技術を支える重要なインフラとなっている。大型計算機システムは、第4期中長期計画にて重点化して取り組むとされた「安全性向上等の革新的技術開発によるカーボンニュートラルへの貢献」、「原子力科学技術に係る多様な研究開発の推進によるイノベーションの創出」、「東京電力福島第一原子力発電所事故の対処に係わる研究開発の推進」、「高レベル放射性廃棄物の処理処分に関する技術開発の着実な実施」、「原子力安全規制行政及び原子力防災に対する支援とそのための安全研究の推進」等といった研究開発活動に利用された。本報告は、令和5年度における大型計算機システムを利用した研究開発の成果を中心に、それを支える利用支援、利用実績、システムの概要等をまとめたものである。
山口 正剛; 海老原 健一; 板倉 充洋; 都留 智仁
Scripta Materialia, 255, p.116366_1 - 116366_5, 2025/01
被引用回数:0 パーセンタイル:0.00(Nanoscience & Nanotechnology)鉄鋼やアルミニウム合金の粒界破壊の原因候補の一つとして水素がもたらす粒界凝集エネルギー低下が考えられている。最近はそれに対する粒界偏析元素の影響が第一原理計算により調べられているが、粒界凝集エネルギーを定量的に評価した研究はない。本研究では、第一原理計算結果を利用した定量的評価手法について述べ、いくつかのテスト計算の例を示す。
山口 瑛子; 高橋 嘉夫*; 奥村 雅彦
Isotope News, (796), p.21 - 23, 2024/12
粘土鉱物は土壌中に豊富に存在し、多くの陽イオンを吸着することから様々な元素の環境動態を支配している。粘土鉱物の吸着の強さは分子レベルの吸着構造によって異なるため、分子レベルの吸着構造が何によって決定するのか、系統的な理解が重要である。本研究では、広域X線吸収微細構造(EXAFS)測定と第一原理シミュレーションを用いて、ラジウムをはじめとした多くの陽イオンの吸着構造を系統的に解明した。その結果、吸着構造の決定には吸着イオンの大きさと水和エンタルピーが重要であるということを示した。
藤澤 唯太*; Krishnadas, A*; Nakamura, Tomonori*; Hsu, C.-H.*; Smith, B. R. M.*; Pardo-Almanza, M*; Hiyane, Hoshu*; 永井 佑紀; 町田 理*; 岡田 佳憲*
Physical Review B, 110(22), p.224511_1 - 224511_5, 2024/12
被引用回数:0 パーセンタイル:0.00(Materials Science, Multidisciplinary)エキゾチックな超伝導体の超伝導磁束の振る舞いを明らかにすることは、今後のデバイス開発にとって重要である。超伝導体LiTi O
O (111)の薄膜を作製し、磁場中での電子状態を走査型トンネル顕微鏡によって観測した。その際、接合部分の曲率に応じて超伝導磁束の状態が大きく変化することがわかった。そして、その変化は理論計算によっても確かめられた。この結果は、Josephson磁束を実験的な構造変化によってコントロールできる可能性を示唆しており、今後の新たなデバイス開発において重要な結果となるだろう。
(111)の薄膜を作製し、磁場中での電子状態を走査型トンネル顕微鏡によって観測した。その際、接合部分の曲率に応じて超伝導磁束の状態が大きく変化することがわかった。そして、その変化は理論計算によっても確かめられた。この結果は、Josephson磁束を実験的な構造変化によってコントロールできる可能性を示唆しており、今後の新たなデバイス開発において重要な結果となるだろう。
岡田 和歩*; 柴田 曉伸*; 木村 勇次*; 山口 正剛; 海老原 健一; 辻 伸泰*
Acta Materialia, 280, p.120288_1 - 120288_14, 2024/11
被引用回数:2 パーセンタイル:36.18(Materials Science, Multidisciplinary)The present study aimed at strengthening prior austenite grain boundary (PAGB) cohesive energy using carbon segregation and investigated the effect of carbon segregation at PAGB on the microscopic crack propagation behavior of hydrogen-related intergranular fractures in high-strength martensitic steels. At the low hydrogen content (below 0.2 wt. ppm), the fracture initiation toughness (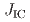 ) and tearing modulus (
) and tearing modulus (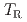 ), corresponding to crack growth resistance, were significantly improved by carbon segregation. In contrast, and did not change by carbon segregation at the high hydrogen content (above 0.5 wt. ppm). Considering the non-linear relationship between the toughness properties and the PAGB cohesive energy, the experimentally evaluated toughness properties ( and ) and the GB cohesive energy previously calculated by first-principles calculations were semi-quantitatively consistent even at the high hydrogen content. The microstructure observation confirmed that the plastic deformation associated with crack propagation, such as the local ductile fracture of uncracked ligaments and the formation of dislocation cell structures/nano-voids, played an important role in the non-linear relationship between the toughness properties and PAGB cohesive energy.
), corresponding to crack growth resistance, were significantly improved by carbon segregation. In contrast, and did not change by carbon segregation at the high hydrogen content (above 0.5 wt. ppm). Considering the non-linear relationship between the toughness properties and the PAGB cohesive energy, the experimentally evaluated toughness properties ( and ) and the GB cohesive energy previously calculated by first-principles calculations were semi-quantitatively consistent even at the high hydrogen content. The microstructure observation confirmed that the plastic deformation associated with crack propagation, such as the local ductile fracture of uncracked ligaments and the formation of dislocation cell structures/nano-voids, played an important role in the non-linear relationship between the toughness properties and PAGB cohesive energy.
 and machine learning potentials for modeling nuclear quantum effects in water
and machine learning potentials for modeling nuclear quantum effects in waterThomsen, B.; 永井 佑紀*; 小林 恵太; 濱田 幾太郎*; 志賀 基之
Journal of Chemical Physics, 161(20), p.204109_1 - 204109_18, 2024/11
被引用回数:1 パーセンタイル:37.13(Chemistry, Physical)SL-PIHMC-MIX法は、第一原理計算と機械学習ポテンシャルを混合した自己学習型経路積分ハイブリッドモンテカルロ法である。SL-PIHMC-MIX法を用いると、第一原理経路積分分子動力学法(PIMD法)よりも、室温の水の構造を計算し収束させるのに必要な第一原理DFT計算の回数を一桁減らすことができる。
永井 佑紀; 富谷 昭夫*
Journal of the Physical Society of Japan, 93(11), p.114007_1 - 114007_8, 2024/11
被引用回数:1 パーセンタイル:43.84(Physics, Multidisciplinary)生成AIにおける基幹技術であるTransformerを用いた新しい自己学習モンテカルロ法を提案した。本研究では、文章における距離の離れた単語の関連性を推測することができるTransformerのAttention機構を用いることにより、電子系の相転移で重要となる長距離相関を効率よく取り込める有効模型を構築した。さらに、スピン回転、空間並進など系が満たすべき対称性をネットワークに取り込むことにより、パラメータ数を劇的に減らすことに成功した。また、レイヤー数を増やすにつれてlossが減っていくというスケーリング則を見出した。
Catumba, G.*; 平口 敦基; Hou, W.-S.*; Jansen, K.*; Kao, Y.-J.*; David Lin, C.-J.*; Ramos, A.*; Sarkar, M.*
Physical Review Research (Internet), 6(4), p.043172_1 - 043172_12, 2024/11
様々な表現の物質場と結合するゲージ理論は、物理学の様々な分野で重要な役割を果たしている。最近、銅酸化物超伝導体の最適ドーピング近傍の興味深い擬ギャップ相のいくつかの側面が、発現したSU(2)ゲージ対称性によって説明できるかもしれないことが提案された。ホールドーピングによる転移付近では、4つの随伴スカラー場と結合した(2+1)次元SU(2)ゲージ理論を構築することができ、異なる破れた対称性を持つ様々な相が存在する豊かな相図を与える。我々は、ハイブリッドモンテカルロ法を用いて、ユークリッド格子上でこのモデルの相図を研究した。その結果、これまでの平均場の研究で予言されていたように、対称性が破れた複数の相が存在することがわかった。4点相互作用によって、摂動論の範囲ではこの理論のSU(2)ゲージ対称性はU(1)か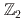 に分解される。さらに、我々はこの理論における閉じ込め-非閉じ込め転移を評価し、我々が研究した格子体積の範囲では、どちらの破れた相も非閉じ込め相であることを見いだした。しかしながら、ポリヤコフループの振る舞いには2つの相で顕著な違いがあることがわかった。
に分解される。さらに、我々はこの理論における閉じ込め-非閉じ込め転移を評価し、我々が研究した格子体積の範囲では、どちらの破れた相も非閉じ込め相であることを見いだした。しかしながら、ポリヤコフループの振る舞いには2つの相で顕著な違いがあることがわかった。
中村 博樹; 町田 昌彦
Proceedings of 31st International Conference on Nuclear Engineering (ICONE31) (Internet), 5 Pages, 2024/11
核燃料開発における安全性の向上のためには、二酸化プルトニウムの熱物性に対する詳細な理解が必要となる。これらの物性を高温で測定することは実験的に困難であるため、密度汎関数理論(DFT)などの数値シミュレーションによって補完する必要がある。PuO の非磁性絶縁基底状態の再現はDFTでは難しかったが、DFT+Uを用いることで非磁性絶縁状態を再現することができた。しかし、得られた状態は磁性状態よりも不安定であり、基底状態とはならなかった。高次相関と厳密な交換エネルギーを取り扱える断熱接続揺動散逸理論(ACFDT)は、この問題に対処する有望な解決策と期待できる。本研究では、ランダム位相近似を用いたACFDTを用いて基底状態のエネルギーを評価し、非磁性状態が磁性状態よりも安定となることを発見した。結果として、PuOの観測された非磁性基底状態の再現に成功したことを意味する。この成果が核燃料材料の熱物性の予測精度の向上に貢献することが期待できる。
の非磁性絶縁基底状態の再現はDFTでは難しかったが、DFT+Uを用いることで非磁性絶縁状態を再現することができた。しかし、得られた状態は磁性状態よりも不安定であり、基底状態とはならなかった。高次相関と厳密な交換エネルギーを取り扱える断熱接続揺動散逸理論(ACFDT)は、この問題に対処する有望な解決策と期待できる。本研究では、ランダム位相近似を用いたACFDTを用いて基底状態のエネルギーを評価し、非磁性状態が磁性状態よりも安定となることを発見した。結果として、PuOの観測された非磁性基底状態の再現に成功したことを意味する。この成果が核燃料材料の熱物性の予測精度の向上に貢献することが期待できる。
Catumba, G.*; 平口 敦基; W.-S. Hou, G.*; Jansen, K.*; Kao, Y.-J.*; David Lin, C.-J.*; Ramos, A.*; Sarkar, M.*
Proceedings of Science (Internet), 453, p.362_1 - 362_7, 2024/11
本研究では、最近Sachdevらによって最適ドーピング付近の銅酸化物超伝導体の物理を説明するために提案された、随伴表現の4つのヒッグス場を持つ3次元SU(2)ゲージ理論を議論する。この理論の閉じ込め相は通常のフェルミ液体相に対応し、ヒッグス相は銅酸化物の擬ギャップ相に対応しており、我々はハイブリッドモンテカルロ法を用いて理論の相図を調査した。我々は、先行研究の平均場での計算に定性的に従う様々な相の存在を発見し、銅酸化物におけるそれらの役割について議論する。さらに、閉じ込め非閉じ込め相転移を調べるためにポリヤコフループの振る舞いを調べ、ヒッグス相が安定な非閉じ込め相を持つことを見いだした。
富谷 昭夫*; 永井 佑紀
Proceedings of Science (Internet), 453, p.001_1 - 001_7, 2024/11
機械学習、ディープラーニングは、格子系の計算物理学を加速させてきた。対称性に対する同変性は、機械学習モデルによって記述される確率分布に強い帰納バイアスを課すため、物理系のシミュレーションには不可欠である。しかし、モデルに対称性を課すことは、自己学習モンテカルロ法(SLMC)において、時に低いアクセプト率を引き起こす。一方、GPTのようなトランスフォーマーで用いられるアテンション機構は、大きなモデルキャパシティを実現する。そこで、我々は、対称性に対する同変性を持ったアテンション機構をSLMCに導入する。我々のアーキテクチャを評価するために、2次元格子上のスピン-フェルミオンモデルに適用を行った。その結果、線形有効モデルを使ったSLMCのアクセプト率を改善し、アクセプト率のスケーリング則を観測した。
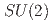 gauge fields
gauge fieldsCatumba, G.*; 平口 敦基; W.-S. Hou, G.*; Jansen, K.*; Kao, Y.-J.*; David Lin, C.-J.*; Ramos, A.*; Sarkar, M.*
Proceedings of Science (Internet), 453, p.87_1 - 87_9, 2024/11
本研究では、ゲージ場と相互作用する一般的な2ヒッグスダブレット模型を格子ゲージ理論で議論する。秩序変数の代わりとしてゲージ不変の大域的観測量を計算することにより、この模型の相図を調べた。それぞれの相において、裸の結合定数の組み合わせや対称性の破れのパターンを変えて理論のスカラー粒子およびベクトルボソン粒子の質量の評価を行なった。またスケール設定と走るゲージ結合定数の決定は、ウィルソンフロウの計算によって行なった。
宇野 功一郎*; 奥村 雅彦; 中尾 淳*; 山口 瑛子; 矢内 純太*
Science of the Total Environment, 949, p.175012_1 - 175012_8, 2024/11
被引用回数:0 パーセンタイル:0.00(Environmental Sciences)雲母鉱物が部分的に風化して生成するフレイドエッジサイト(FES)は、Csイオンを選択的に吸着するが、その詳細なメカニズムは未解明であった。これを明らかにするため、本研究では、雲母の陽イオン抽出とCs吸着実験を行い、ルビジウムを吸着した雲母よりもカリウムを吸着した雲母の方がCsを多く吸着することがわかった。この原因を明らかにするため、セシウム、ルビジウム、カリウムがFESに吸着した構造の安定性を第一原理計算により評価した結果、Cs吸着前に存在する陽イオン種がカリウムであることがCsの安定性に重要であることがわかった。
