


Li, H.*; 櫻庭 俊; Chandrasekaran, A.*; Yang, L.-W.*
Li, H.*; Sakuraba, Shun; Chandrasekaran, A.*; Yang, L.-W.*
本論文では、タンパク質-タンパク質並びにタンパク質-リガンド結合部位がタンパク質の形状と、内在的なタンパク質運動に強く依存することを示す。68のタンパク質-タンパク質複合体ならびに240の非配列相同的な酵素を対象に解析した結果、結合部位は分子振動が最も小さくなる場所に位置しやすいこと、またタンパク質-タンパク質複合体の場合は各タンパク質の回転・曲げ変形角が最大となるよう位置することを発見した。さらに、この発見をタンパク質ドッキング計算に応用することで、結合位置が天然構造に一致する率を2倍に引き上げた。また、酵素を対象とした場合、90%の活性残基がタンパク質の内在性ドメインの境界面に近い50%の残基に見いだされることを発見した。これらの結果は酵素の活性部位が重心から見て非等方的に位置することを示唆した。
We provide evidence supporting that protein-protein and protein-ligand docking poses are functions of protein shape and intrinsic dynamics. Over sets of 68 protein-protein complexes and 240 non-homologous enzymes, we recognize common predispositions for binding sites to have minimal vibrations and angular momenta while two interacting proteins orient so as to maximize the angle between their rotation/bending axes (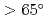 ). The findings are then used to define quantitative criteria to filter out docking decoys less likely to be the near-native poses, hence the chances to find near-native hits can be doubled. With the novel approach to partition a protein into "domains" of robust but disparate intrinsic dynamics, 90% of catalytic residues in enzymes can be found within the first 50% of the residues closest to the interface of these dynamics domains. The results suggest an anisotropic rather than isotropic distribution of catalytic residues near the mass centers of enzymes.
). The findings are then used to define quantitative criteria to filter out docking decoys less likely to be the near-native poses, hence the chances to find near-native hits can be doubled. With the novel approach to partition a protein into "domains" of robust but disparate intrinsic dynamics, 90% of catalytic residues in enzymes can be found within the first 50% of the residues closest to the interface of these dynamics domains. The results suggest an anisotropic rather than isotropic distribution of catalytic residues near the mass centers of enzymes.
発表言語 |
: | English |
|---|---|---|
掲載資料名 |
: | |
巻 |
: | 54 |
号 |
: | 8 |
ページ数 |
: | p.2275 - 2285 |
発行年月 |
: | 2014/08 |
キーワード |
: | no keyword |
論文URL |
: |
|
|---|---|---|
論文解説記事 |
: |
Access |
: |
- Accesses |
|---|---|---|
InCites™ |
: |
パーセンタイル:69.13 分野:Chemistry, Medicinal |
Altmetrics |
: |
[CLARIVATE ANALYTICS], [WEB OF SCIENCE], [HIGHLY CITED PAPER & CUP LOGO] and [HOT PAPER & FIRE LOGO] are trademarks of Clarivate Analytics, and/or its affiliated company or companies, and used herein by permission and/or license.
