


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
池部 仁善; 桜庭 俊*; 河野 秀俊
PLOS Computational Biology, 12(3), p.e1004788_1 - e1004788_13, 2016/03
被引用回数:50 パーセンタイル:90.68(Biochemical Research Methods)Acetylation of lysine residues in histone tails is associated with gene transcription. Because histone tails are structurally flexible and intrinsically disordered, it is difficult to experimentally determine the tail conformations and the impact of acetylation. In this work, we performed simulations to sample H3 tail conformations with and without acetylation. The results show that irrespective of the presence or absence of the acetylation, the H3 tail remains in contact with the DNA and assumes an alpha-helix structure in some regions. Acetylation slightly weakened the interaction between the tail and DNA and enhanced alpha-helix formation, resulting in a more compact tail conformation. We inferred that this compaction induces unwrapping and exposure of the linker DNA, enabling DNA-binding proteins (e.g., transcription factors) to bind to their target sequences. In addition, our simulation also showed that acetylated lysine was more often exposed to the solvent, which is consistent with the fact that acetylation functions as a post-translational modification recognition site marker.
櫻庭 俊
統計数理, 62(2), p.171 - 184, 2014/12
生体分子の分子シミュレーションは、大量の時系列データを出力するため、そのデータ量の削減が極めて重要になる。本稿では、分子シミュレーション分野で用いられている線形変換に基づく様々な次元削減手法と、そこに現れる相関の考え方を扱う。実例として機能モード分析、主成分分析、全相関分析、準非調和分析、独立部分空間分析などを紹介する。また、その物理学的な意味についても解説を行う。
Li, H.*; 櫻庭 俊; Chandrasekaran, A.*; Yang, L.-W.*
Journal of Chemical Information and Modeling, 54(8), p.2275 - 2285, 2014/08
被引用回数:17 パーセンタイル:69.13(Chemistry, Medicinal)本論文では、タンパク質-タンパク質並びにタンパク質-リガンド結合部位がタンパク質の形状と、内在的なタンパク質運動に強く依存することを示す。68のタンパク質-タンパク質複合体ならびに240の非配列相同的な酵素を対象に解析した結果、結合部位は分子振動が最も小さくなる場所に位置しやすいこと、またタンパク質-タンパク質複合体の場合は各タンパク質の回転・曲げ変形角が最大となるよう位置することを発見した。さらに、この発見をタンパク質ドッキング計算に応用することで、結合位置が天然構造に一致する率を2倍に引き上げた。また、酵素を対象とした場合、90%の活性残基がタンパク質の内在性ドメインの境界面に近い50%の残基に見いだされることを発見した。これらの結果は酵素の活性部位が重心から見て非等方的に位置することを示唆した。
櫻庭 俊; 松林 伸幸*
Journal of Computational Chemistry, 35(21), p.1592 - 1608, 2014/08
被引用回数:61 パーセンタイル:80.66(Chemistry, Multidisciplinary)ERmodはエネルギー表示法に基づく、効率よく溶媒和自由エネルギーを近似計算するソフトウェアである。溶液系・参照溶媒系の2状態での分子シミュレーションのみから、溶媒和自由エネルギーを計算することが可能である。ERmodのサブプログラムである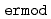 は溶質溶媒間相互作用エネルギーの分布関数を計算し、別のサブプログラムである
は溶質溶媒間相互作用エネルギーの分布関数を計算し、別のサブプログラムである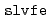 は溶媒和自由エネルギーを分布関数から溶液の近似汎関数理論に従い計算する。本論文ではERmodの設計と実装について記述し、またそのパフォーマンスを2つの有機分子と2つのタンパク質を溶質とする系について示す。ERmodの利用範囲は幅広く、均一溶媒系である流体やポリマーのみならず、ミセルや脂質膜系などのナノ不均一系まで適用可能である。ERmodは
は溶媒和自由エネルギーを分布関数から溶液の近似汎関数理論に従い計算する。本論文ではERmodの設計と実装について記述し、またそのパフォーマンスを2つの有機分子と2つのタンパク質を溶質とする系について示す。ERmodの利用範囲は幅広く、均一溶媒系である流体やポリマーのみならず、ミセルや脂質膜系などのナノ不均一系まで適用可能である。ERmodは にて公開されている。
にて公開されている。
池部 仁善; 櫻庭 俊; 河野 秀俊
Journal of Computational Chemistry, 35(1), p.39 - 50, 2014/01
被引用回数:17 パーセンタイル:44.03(Chemistry, Multidisciplinary)A novel, efficient sampling method for biomolecules is proposed. The partial McMD was recently developed as a method that improved generalized ensemble (GE) methods to focus sampling only on a part of a system (GEPS); however, it was not tested well. We found that partial McMD did not work well for poly-lysine decapeptide and gave significantly worse sampling efficiency than a conventional GE. Herein, we elucidate the fundamental reason for this and propose a novel GEPS, adaptive lambda square dynamics (ALSD), which can resolve the problem faced when using partial McMD. We demonstrate that ALSD greatly increases the sampling efficiency over a conventional GE. We believe that ALSD is an effective method and is applicable to the conformational sampling of larger and more complicated biomolecule systems.
河野 秀俊; 池部 仁善; 桜庭 俊*; 石田 恒
no journal, ,
Dynamics of the nucleosome and accessibility of nucleosomal DNA play critical roles in many nuclear processes. Nucleosomes themselves are highly dynamic and the nucleosomal DNA is spontaneously unwrapped to make it accessible to various DNA-binding proteins. Histone variants are known for each of histone proteins and are supposed to have different roles in the regulation of nuclear processes. In addition, post-translational modifications of histones have been known to affect the regulation, but it is still unknown how they affect changes of the structure and dynamics of chromatin. Through computer simulations, we are investigating the following topics to understand the dynamics of nucleosomes: (1) how much it costs for unwrapping nucleosomal DNA from the histone core, (2) how different the dynamics are among nucleosomes composed of histone variants and (3) how acetylation of histone tails impacts on nucleosome structures. Our simulations suggest that the cost for unwrapping 1bp of DNA from histone core is 0.1 to 0.4 kcal/mol and it changes according to nucleosome types. In the dissociation of nucleosomal DNA, asymmetric unwrapping (unwrapping either end of DNA) became remarkable when the DNA unwrapping reached the inner turn, probably because DNA-DNA repulsion disappeared at this stage, thereby histone-DNA interactions were stabilized. Acetylation of K14 of H3 tail enhanced the DNA flexibility.
池部 仁善; 桜庭 俊*; 河野 秀俊
no journal, ,
In eukaryotic cells, genome DNA forms nucleosomes composed of DNA wrapped around a histone octamer: two copies of H3, H4, H2A, and H2B histone proteins, and it is compactly stored in the nucleus. It is known that acetylation of lysine residues on H3 and H4 histone tails, which are N-terminal regions of H3 and H4 histone proteins, generally induces transcription. To elucidate the mechanism of the regulation, we performed conformational sampling of an acetylated H3 histone tail with Adaptive Lambda Square Dynamics (ALSD) simulation and compared the result with our previous simulation result of unacetylated H3 histone tail system. The results showed that H3 histone tail were almost always located nearby DNA regardless of the acetylation, which is against the conventional view that the acetylation is expected to cause major structural changes. However, the acetylation induced reduction of interactions between the histone tail and DNA and enhanced alpha helix formation of the histone tail. We infer that the histone tail compaction caused by the alpha helix formation induces unwrapping of DNA at entry/exit regions more and increases the chance of DNA-binding proteins bind to DNA. These results give a new view of how acetylation affects chromatin conformation.
池部 仁善; 桜庭 俊*; 河野 秀俊
no journal, ,
In eukaryotic cells, genome DNA forms nucleosomes composed of DNA wrapped around a histone octamer: two copies of H3, H4, H2A, and H2B histone proteins, and it is compactly stored in the nucleus. It is known that acetylation of lysine residues on H3 and H4 histone tails, which are N-terminal regions of H3 and H4 histone proteins, generally induces transcription. To elucidate the mechanism of the regulation, we performed conformational sampling of an acetylated H3 histone tail with Adaptive Lambda Square Dynamics (ALSD) simulation and compared the result with our previous simulation result of unacetylated H3 histone tail system. The results showed that H3 histone tail were almost always located nearby DNA regardless of the acetylation, which is against the conventional view that the acetylation is expected to cause major structural changes. However, the acetylation induced reduction of interactions between the histone tail and DNA and enhanced alpha helix formation of the histone tail. We infer that the histone tail compaction caused by the alpha helix formation induces unwrapping of DNA at entry/exit regions more and increases the chance of DNA-binding proteins bind to DNA. These results give a new view of how acetylation affects chromatin conformation.
池部 仁善; Li, Z.; 桜庭 俊*; 石田 恒; 河野 秀俊
no journal, ,
Nucleosome is the fundamental structural unit of chromatin. Histone proteins which constitute the nucleosome have their variants. In addition, histone proteins are subject to post-translational modification (PTM) to function in epigenetics. Acetylation of histone tails generally functions to activate gene expression, though the molecular mechanism is not well understood. We carried out conventional molecular dynamics simulations and enhanced sampling simulation to examine the impact of histone variant and the acetylation on the stability and dynamics of nucleosome. The results suggest that (1) stability of nucleosome depends on constituted histones; (2) N-terminal tail of H3 and C-terminal tail of H2A are both associated with dynamics of liker DNA and (3) acetylation makes the H3 tail conformation more compact and enhances dissociation of nucleosomal DNA from the histone core. Further, the acetylated lysine was more exposed to the solvent, which is consistent with its role as a PTM recognition site marker. These findings increase our understanding of role of histone variants and the impact of PTM on nucleosome stability and dynamics and on the higher order structure of chromatin.
河野 秀俊; 池部 仁善; Li, Z.; 桜庭 俊*
no journal, ,
ヌクレオソームを構成しているヒストンタンパク質は、ヒストンテールと呼ばれる一定の構造を取らない領域(天然変性領域)が存在する。テールは、翻訳後修飾をうけ、エピジェネティクスマーカーとしての役割を果たしている。ヌクレオソームのコア領域は、X線結晶構造解析によりその立体構造が明らかにされているものの、テール領域は天然変性状態にあるため立体構造は依然として分かっていない。また、翻訳後修飾がテールの構造にどのような影響を与えるのかは依然として謎のままである。そこで、我々はコンピュータシミュレーションによってテールの構造を調べている。今回は、ヒストンH3のN末テール及びH2AのC末テールの役割について報告する。H3テールは、テールの中でも最もよく研究されている対象であり、アセチル化は基本的に転写を活性化することが報告されている。アセチル化がヌクレオソーム構造に与える影響を調べるために、アセチル化ありなしの状態でのH3テールの構造を調べた。これまでのシミュレーション計算では、テール部分のみを切り出した計算しかなされてこなかったが、今回初めてヌクレオソーム存在化でのテールの構造を明らかにした。計算の結果、アセチル化ありなしに関わらずテールはDNAに張り付いた構造をとること、テールのアルギニンは80%以上の確率でDNAと相互作用しているがリジンは適度に溶媒に露出すること、テールの位置によってリンカーDNAの開き具合がかわること、アセチル化によってテールのへリックス構造形成率があがることなどが分かった。これらを統合して考えると、H3テールのアセチル化によってテールがコンパクトな構造をとり、転写活性を促すようにリンカーDNAの束縛を緩めてヒストンコアから開かせる効果があることが分かった。また、この効果はアセチル化が蓄積すればするほど大きいことが分かった。一方、H2AのC末テールは生化学的にはあまり研究されていない。我々のシミュレーション結果は、H2AのC末テールのダイナミックな動き、つまり、テールのインナーDNAからリンカーDNAへのジャンプがリンカーDNAの開閉と高い相関があることを見出した。H2AのC末テールを短くしたシミュレーションではリンカーDNAが開くことなどから、H2AのC末テールはリンカーDNAを閉じた状態を安定化していることが示唆された。
池部 仁善; 櫻庭 俊; 河野 秀俊
no journal, ,
A novel, efficient sampling method for biomolecules is proposed. The partial multicanonical molecular dynamics (partial McMD) was recently developed as a generalized ensemble (GE) method to focus sampling only on a part of a system (GEPS); however, it was not tested well. We found that partial McMD did not work well for poly-lysine decapeptide and gave significantly worse sampling efficiency than a conventional GE. Herein, we elucidate the fundamental reason for this and propose a novel GEPS, adaptive lambda square dynamics (ALSD), which can resolve the problem faced when using partial McMD. We demonstrate that ALSD greatly increases the sampling efficiency over a conventional GE.
池部 仁善; 櫻庭 俊; 河野 秀俊
no journal, ,
A novel, efficient sampling method for biomolecules is proposed. The partial Multicanonical Molecular Dynamics (partial McMD, Okumura (2008)) simulation was recently developed as an improved generalized ensemble (GE) methods to focus sampling only on a part of a system (GEPS); he expected that the focused sampling reduced the energy space to be sampled and concomitantly increased the efficiency of the conformational sampling. However, the partial McMD has not been tested well except for an alanine dipeptide system. We found that partial McMD did not work well for poly-lysine decapeptide and gave significantly worse sampling efficiency than a conventional GE. Herein, we elucidate the fundamental reason for this and propose a novel GEPS, adaptive lambda square dynamics (ALSD), which can resolve the problem faced when using partial McMD. We demonstrate that ALSD greatly increases the sampling efficiency of the peptide conformation over a conventional GE by scaling of a partial potential energy: electrostatic, van der Waals, and torsion energies relevant to the peptide. We believe that ALSD is an effective method and is applicable to the conformational sampling of larger and more complicated biomolecule systems. Furthermore, ALSD can be available for conformational sampling of biomolecules with intrinsically disordered regions.
池部 仁善; 櫻庭 俊; 河野 秀俊
no journal, ,
真核生物のDNAはクロマチン構造を形成することで核内にコンパクトに収納されているが、DNA機能発現の際にはこの構造は一時的に解きほぐされる。この構造の解体と再形成のメカニズムはDNA機能制御において重要であり、クロマチンを構成する要素の一つであるヒストンテールへの化学修飾によりコントロールされることが知られている。しかし天然変性蛋白領域(Intrinsically Disordered Region: IDR)であるテールの構造を実験的に調査することは難しく、これまでそのメカニズムの詳細は明らかとなっていなかった。このメカニズムを解明するための第一段階として、我々はまず化学修飾なしのテールの状態を明らかにすべく、全原子モデルを用いたシミュレーションを行っている。我々は当初、partial McMDと呼ばれるシミュレーション手法を用いて計算を行っていたが、その計算の過程で本手法が抱える問題点を発見した。前回の発表ではpartial McMDの問題点とその原因について発表を行った。今回の発表では、partial McMDが抱える問題点を解決すべく我々が新たに開発した計算手法、Adaptive Lambda Square Dynamics(ALSD)法について紹介し、本手法を用いたH3ヒストンテールの構造探索計算の結果を発表する。
池部 仁善; 櫻庭 俊; 河野 秀俊
no journal, ,
多数の正電荷アミノ酸(リジンとアルギニン)を含むヒストンテールは、クロマチン形成において、DNA由来の強い負電荷を持つヌクレオソーム同士の凝集を仲介することが知られている。また、ヒストンテールは化学修飾のターゲットでもあり、エピゲノムのマーカーとしての重要性から多くの研究がなされている。数ある化学修飾の中でも、リジン残基のアセチル化は転写を活性化する代表的なマーカーとして知られる。リジンの正電荷を中性化するアセチル化修飾は、DNAとヒストンテール間の引力相互作用を減少させる。その結果クロマチンの凝集が緩み、転写因子がDNAに結合できるようになることで転写が活性化される、という転写制御メカニズムが提唱されているが、その詳細は未だ明らかになっていない。そこで我々は、我々が新たに開発した、ヒストンテールのとりうる立体構造を効率的に探索する分子動力学シミュレーション手法、adaptive lambda square dynamics (ALSD)法によって、2パターンのH3ヒストンテール系[(1)修飾無しの系、(2)14残基目のリジンがアセチル化(K14Ac)された系]がどのような立体構造集団を形成するのかを明らかにした。本発表では、2つの構造集団の比較から、ヒストンテールのアセチル化がその立体構造に与える影響について紹介する。
河野 秀俊; 櫻庭 俊; 石田 恒
no journal, ,
Eukaryotic genome is compactly stored into a tiny nucleus of cell in a form of protein-DNA complex. This protein-DNA complex is called as nucleosome which is composed of a histone octamer formed by two copies each of the four core histones H3, H4, H2A and H2B and about 150bp of DNA wrapping almost twice around the octamer. This compact form, however, has to be unwrapped in transcription, DNA duplication and DNA repair processes. To study the detailed molecular mechanism, we calculated free energy profiles for unwrapping nucleosomal DNA on H3-containg and CENP-A containing nucleosomes with adaptively biased MD and umbrella sampling. We estimated from the profiles that a cost for unwrapping the outer DNA is 0.1 to 0.4 kcal/mol/1bp, consistent with a value experimentally obtained. Compared with H3 nucleosome, the profile showed that CENP-A nucleosome was the most stable in a conformation where 15bp from each of ends were unwrapped. The detail analysis indicated that this is attributable to the difference in ability of forming hydrogen bond of alphaN helix of H3 and CENP-A.
池部 仁善; 櫻庭 俊; 河野 秀俊
no journal, ,
多数の正電荷アミノ酸(リジンとアルギニン)を含むヒストンテールは、クロマチン形成において、DNA由来の強い負電荷を持つヌクレオソーム同士の凝集を仲介することが知られている。また、ヒストンテールは化学修飾のターゲットでもあり、エピゲノムのマーカーとしての重要性から多くの研究がなされている。数ある化学修飾 の中でも、リジン残基のアセチル化は転写を活性化する代表的なマーカーとして知られる。リジンの正電荷を中性 化するアセチル化修飾は、DNAとヒストンテール間の引力相互作用を減少させ、その結果クロマチンの凝集が緩み、転写因子がDNAに結合できるようになることで転写が活性化される、という転写制御メカニズムが提唱されているが、その詳細は未だ明らかになっていない。そこで我々は、新たに開発した、ヒストンテールのとりうる立体構造を効率的に探索する分子動力学シミュレーション手法、adaptive lambda square dynamics (ALSD)法 によって、2パターンのH3ヒストンテール系(1: 修飾無しの系、2: 14残基目のリジンがアセチル化(K14Ac)された系)がどのような立体構造集団を形成するのかを明らかにした。本発表では、2つの構造集団の比較から、ヒストン テールのアセチル化がその立体構造に与える影響について紹介する。
池部 仁善; 桜庭 俊*; 河野 秀俊
no journal, ,
In eukaryotic cells, genome DNA is stored in a complex with histone proteins (H3, H4, H2A and H2B). Acetylation to terminal regions of histones (histone tails) is generally believed to regulate gene expression through dissociation of tails from DNA, although conformations of disordered tails remain poorly understood. In this work, we examined differences in conformational ensembles of H3 tail with or without K14 acetylation using adaptive lambda square dynamics simulation. The result suggested that the acetylation does not make the tail dissociate from DNA. Instead, it enhanced secondary structure formation of the tail and unwrapping of DNA from the structured histone core regions. This study elucidated the first step of the gene regulation mechanism.
河野 秀俊; 池部 仁善; 桜庭 俊*; 石田 恒
no journal, ,
Eukaryotic genome DNA is compactly packaged into a nucleus of cell in a form of protein-DNA complex. This protein-DNA complex is called as nucleosome which is composed of a histone octamer formed by two copies each of the four core histones H3, H4, H2A and H2B and about 150 bp of DNA wrapping almost twice around the octamer. Nucleosome, however, changes its form by replacing histones with their variants and post-translational modifications to function in transcription, DNA duplication and DNA repair processes. To study the impact of histone variants and post-translational modifications on the structure and stability of nucleosome, we have performed molecular dynamics simulations with enhanced sampling methods. Free energy profiles obtained by nucleosomal DNA-unwrapping simulations exhibited that H3-containing nucleosome was stable in a fully wrapped state. It cost about 10 kcal/mol for unwrapping the first 8 bp from the DNA end. The cost much decreased for further unwrapping up to 20 bp-unwrapping. In contrast, CENP-A containing nucleosome was the most stable in a state where 5 to 10 bp of DNA were unwrapped from either end of DNA. For both nucleosomes, we observed that DNA was unwrapped asymmetrically. Next we evaluated a post-translational modification, K14 acetylation of H3 histone in a nucleosome structure. Our simulations indicated that H3 histone tail was almost always located nearby DNA regardless of the acetylation, however, K14 acetylation enhanced unwrapping of DNA at entry/exit regions. This can be explained by reduction of interactions between the histone tail and DNA due to the charge neutralization and increase in alpha-helical content in the histone tail. As a consequence, K14 acetylation increases the chance that DNA-binding proteins access DNA. These results give a new view of how acetylation affects chromatin conformation.
河野 秀俊; 米谷 佳晃; 池部 仁善; 櫻庭 俊; 石田 恒
no journal, ,
Eukaryotic Genome is stored in a nucleus in the form of chromatin. Along with DNA binding proteins, this chromatin structure plays a key role in gene regulation. The chromatin has the fundamental structural unit called nucleosome which is composed of two copies of histones H2A, H2B, H3 and H4 and DNA wrapped around the histone proteins almost twice. Transcription does not occur until nucleosome is unwrapped to expose DNA, which can then be recognized by regulatory proteins. To understand this unwrapping process, MD simulations were performed on two nucleosomes: the canonical H3 and the H3 variant called CENP-A, essential for the kinetochore assembly. In the talk, how these nucleosomes are different in free energy profiles and what causes the differences are discussed.
 イオン-タンパク質系の古典分子動力学計算
イオン-タンパク質系の古典分子動力学計算櫻庭 俊
no journal, ,
福島第一原子力発電所の事故以降、環境中に放出された放射性降下物が問題となっている。中でも放射性セシウム(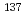 Cs)の分布とその生物学的な濃縮は国内で大きな関心を呼んでいる。Csイオンの生化学的な挙動、たとえばタンパク質との結合の挙動などをシミュレーションから調査するためには、またセシウム吸着高分子の設計などを行うためには、溶液中・タンパク質近傍のCsイオンの挙動を忠実に再現する、適切な力場を用意する必要がある。本発表では、現在知られているタンパク質用の古典動力学用の力場と互換性のあるCs
Cs)の分布とその生物学的な濃縮は国内で大きな関心を呼んでいる。Csイオンの生化学的な挙動、たとえばタンパク質との結合の挙動などをシミュレーションから調査するためには、またセシウム吸着高分子の設計などを行うためには、溶液中・タンパク質近傍のCsイオンの挙動を忠実に再現する、適切な力場を用意する必要がある。本発表では、現在知られているタンパク質用の古典動力学用の力場と互換性のあるCs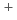 イオン力場に対するベンチマークを行った結果を報告する。また、国内外の研究では、Csイオン-
イオン力場に対するベンチマークを行った結果を報告する。また、国内外の研究では、Csイオン-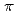 電子相互作用がCsイオン-タンパク質の相互作用に重要であることが指摘されている。本研究では、上記のベンチマークに合わせて、Csイオン-電子相互作用を既存パラメータに対し補整項として導入することを試み、結果を報告する。
電子相互作用がCsイオン-タンパク質の相互作用に重要であることが指摘されている。本研究では、上記のベンチマークに合わせて、Csイオン-電子相互作用を既存パラメータに対し補整項として導入することを試み、結果を報告する。
