


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
大沼 敏治*; 宮下 敦巳; 吉川 正人; 土田 秀一*; 岩沢 美佐子*
平成21年度先端研究施設共用促進事業「地球シミュレータ産業戦略利用プログラム」利用成果報告書, p.21 - 27, 2010/07
ワイドギャップ半導体である炭化珪素(SiC)は従来のシリコン(Si)半導体に比べて飛躍的な性能向上を実現するパワー半導体デバイスの材料として期待されている。また、SiC半導体デバイスは低損失の省エネデバイスとして開発が進められているとともに、Si半導体デバイスと同様に熱酸化により酸化絶縁膜を作製できるため、次世代のMOS型パワーデバイスとして有望である。しかし、従来のSiC MOS型パワーデバイスは、界面トラップの存在等によりチャネル移動度が理論的な予想値より遥かに小さく、優れた特性を発揮できていなかった。これらの特性を改善するためには、原子レベルで界面の構造と熱酸化の機構を明らかにすることが重要となる。SiCの熱酸化過程のシミュレーションにおいては、化学反応を伴うことと、界面においてさまざまな結合があることから、経験的なパラメータを一切用いない第一原理法が強力なツールとなるが、計算量が膨大なためこれまで行われてこなかった。地球シミュレータによる大規模な第一原理分子動力学計算によりSiCの熱酸化過程・アニーリング及び界面準位のシミュレーションが可能になったのでここに報告する。
 /SiC interface structure by the first-principles molecular dynamics simulation
/SiC interface structure by the first-principles molecular dynamics simulation宮下 敦巳; 大沼 敏治*; 岩沢 美佐子*; 土田 秀一*; 吉川 正人
Materials Science Forum, 556-557, p.521 - 524, 2007/00
SiCデバイスは宇宙や原子炉等の極限環境下で動作する素子として期待されている。しかしながら現状のSiCデバイスは理論的に予想されている性能を発揮しているとは言いがたい。その理由はSiCとその酸化膜であるSiOとの界面に存在する欠陥が素子の性能を低下させているからだと考えられる。実デバイスにある界面欠陥構造を計算機シミュレーションで再現しようとするなら、現実の界面にあるようなアモルファスSiO/SiCの構造を計算機上に再現することが非常に重要となってくる。われわれは444原子からなる結晶/結晶界面構造を計算機上に構築し、それに対して加熱・急冷計算を行うことでアモルファスSiO/SiC構造を生成した。加熱温度,加熱時間,急冷速度はそれぞれ4000K, 3ps, -1000K/psである。得られた界面構造のSiO領域はバルクのアモルファスSiO構造とよく適合し、界面におけるダングリングボンド欠陥も消滅していることが確かめられた。
/4H-SiC(0001) interface oxidation process; From first-principles大沼 敏治*; 宮下 敦巳; 岩沢 美佐子*; 吉川 正人; 土田 秀一*
Materials Science Forum, 556-557, p.615 - 620, 2007/00
平面波近似とスーパーセルモデルを用い、SiO/4H-SiC(0001)酸化過程の第一原理分子動力学計算による動的シミュレーションを行った。反応の初期構造の生成には加熱・急冷法を用いた。この初期構造は界面ダングリングボンドのないSiO/SiC構造である。酸化反応の引き金とするために、界面付近のSiC層に炭素空孔を導入した。酸化反応シミュレーションは界面付近の空隙に酸素分子を一つずつ置いて行くことによって行った。酸化反応シミュレーションは2500Kの下で行った。酸素分子は解離しSiO中のSi原子と結合を組み、また、界面付近にいるSiC層中のSi原子も酸化されSiO層を形成した。界面欠陥の候補の一つと考えられている炭素クラスタ構造が界面に形成され、さらに、酸素分子は炭素クラスターと反応しCO分子を形成した。
木下 幹康*; Geng, H. Y.*; Chen, Y.*; 金田 保則*; 岩沢 美佐子*; 大沼 敏治*; 園田 健*; 安永 和史*; 松村 晶*; 安田 和弘*; et al.
Proceedings of 2006 International Meeting on LWR Fuel Performance (TopFuel 2006) (CD-ROM), p.248 - 254, 2006/10
ニュー・クロスオーバー・プロジェクト(NXO)は、核燃料物質に対する核分裂放射線照射の影響を大学,国立・私立の研究所に渡る横断的な協力研究により検討している。加速器照射実験及び計算科学的手法を使用して、高燃焼度軽水炉燃料ペレットでリム構造を構成する機構を解明するためのシミュレーション研究が実施されている。加速器照射,高エネルギー電子線照射,核分裂エネルギー粒子線及び希ガス原子のイオン注入等が実施されている。照射試料としては、CeOを核燃料の模擬物質として使用した。最初の成果として、高エネルギー電子照射による平面構造形成、及び、高線量の高エネルギー粒子放射による結晶粒微細化に似た表面改質が観察された。第一原理及び分子動力学(MD)計算を用いた解析を実施し、UOとCeOの点欠陥形成エネルギー及び点欠陥を伴ったXe原子の相互作用についての情報が得られた。
/SiC interface宮下 敦巳; 吉川 正人; 叶野 琢磨; 大沼 敏治*; 酒井 高行*; 岩沢 美佐子*; 曽根田 直樹*
Annual Report of the Earth Simulator Center April 2004 - March 2005, p.287 - 291, 2005/12
Siに比べ優れた物理特性を持つSiCを用いた半導体デバイスは、従来のSiやGaAs半導体デバイスでは動作が困難な、原子炉や宇宙環境等、極限環境下で用いられる素子として期待されている。半導体素子界面では原子レベルの欠陥の荷電状態が電気特性を支配しているため、この界面構造を計算機上で模擬し、界面欠陥構造がどのようにデバイス特性に影響するのか導出するため、地球シミュレータを用いた第一原理分子動力学計算で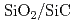 界面構造を構築し電子構造を決定する。400原子程度の中規模モデルを用いてアモルファス界面構造生成を行った。加熱温度は4000K、加熱時間は3ps、急冷速度は-1000K/ps、界面でのSiC可動層は4層とし、2200Kで
界面構造を構築し電子構造を決定する。400原子程度の中規模モデルを用いてアモルファス界面構造生成を行った。加熱温度は4000K、加熱時間は3ps、急冷速度は-1000K/ps、界面でのSiC可動層は4層とし、2200Kで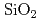 側終端固定層を開放し自由端とすることによって、層でのアモルファス化を促進させた。生成された界面はダングリングボンドが消滅しており、清浄界面に近い状態が再現されたが、バンドギャップ中には欠陥準位が存在するのが観察された。欠陥準位は界面に存在する酸素から生じており、結合に寄与できない局在した電子分布が準位の原因となっていることがわかった。
側終端固定層を開放し自由端とすることによって、層でのアモルファス化を促進させた。生成された界面はダングリングボンドが消滅しており、清浄界面に近い状態が再現されたが、バンドギャップ中には欠陥準位が存在するのが観察された。欠陥準位は界面に存在する酸素から生じており、結合に寄与できない局在した電子分布が準位の原因となっていることがわかった。
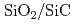 界面の生成
界面の生成宮下 敦巳; 大沼 敏治*; 吉川 正人; 岩沢 美佐子*; 中村 智宣*; 土田 秀一*
no journal, ,
Siに比べ優れた物理特性を持つSiCを用いた半導体デバイスは、従来のSiやGaAs半導体デバイスでは動作が困難な、原子炉や宇宙環境等、極限環境下で用いられる素子として期待されている。しかしながら、SiCの酸化膜とSiCの界面には界面欠陥が多く存在しており、この欠陥構造がSiCデバイスのチャネル移動度低下の原因となっている。半導体素子界面では原子構造が作り出す電子状態がその電気特性に影響を与えることから、界面欠陥構造とデバイス特性との関連性を追求することが重要である。本研究では、デバイス特性に影響を与える欠陥構造の解明を目標とし、欠陥を含んだ実際の界面に近い状態の原子構造を計算機上で模擬しエネルギー準位の計算を行っている。シミュレーション計算には地球シミュレータを用い、第一原理分子動力学計算による加熱・急冷計算によってアモルファス界面構造を構築し電子構造を決定した。シミュレーションには444原子の原子構造モデルを用い、加熱温度4000K、加熱時間3ps、急冷速度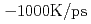 、終端以外の
、終端以外の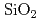 層の可動、界面のSiC可動層4層との条件を用いた。急冷時の2200Kにおいて終端固定層を開放し自由端とすることによって、層でのアモルファス化を促進させた。生成された界面は初期構造では界面Si原子に存在したダングリングボンドが消滅しており、清浄界面に近い状態が再現されていた。しかし、バンドギャップ中にはいまだ準位が存在し、欠陥準位は界面付近に存在する酸素原子の結合に寄与しない局在電荷分布に起因しているこがわかった。
層の可動、界面のSiC可動層4層との条件を用いた。急冷時の2200Kにおいて終端固定層を開放し自由端とすることによって、層でのアモルファス化を促進させた。生成された界面は初期構造では界面Si原子に存在したダングリングボンドが消滅しており、清浄界面に近い状態が再現されていた。しかし、バンドギャップ中にはいまだ準位が存在し、欠陥準位は界面付近に存在する酸素原子の結合に寄与しない局在電荷分布に起因しているこがわかった。
/SiC界面構造の第一原理計算による生成宮下 敦巳; 大沼 敏治*; 岩沢 美佐子*; 吉川 正人; 土田 秀一*
no journal, ,
Siに比べ優れた物理特性を持つSiCを用いた半導体デバイスは、従来のSiやGaAs半導体デバイスでは動作が困難な、原子炉や宇宙環境等,極限環境下で用いられる素子として期待されている。しかしながら、SiCの酸化膜とSiCの界面には、SiCデバイスのチャネル移動度低下の原因となる界面欠陥が多く存在しており、その構造とデバイス特性との関連性を追求することは非常に重要である。本研究では実際の界面に近い状態の原子構造を計算機上で生成し、その電子状態が界面電気特性に与える影響を追求している。実際のデバイス絶縁膜を模擬するために、加熱・急冷計算によりアモルファスSiO/SiC界面構造を計算機上に構築し電子構造を決定した。計算には第一原理分子動力学計算コードであるVASPを用い、地球シミュレータ上で行った。444原子構造モデルを用いた加熱・急冷計算によって、初期構造では界面Si原子に存在していたダングリングボンドが完全に消滅した急峻界面が再現された。この時の条件は、加熱温度4000K,加熱時間3ps,急冷速度-1000K/psであり、さらに、急冷時の2200Kにおいて加熱中は固定してあったSiO終端を自由端とすることによって、SiO層での歪を低減させるとともにアモルファス化を促進させた。バンドギャップ中にはなお欠陥準位が存在したが、これはアモルファスSiO中の酸素のダングリングボンドに起因していることがわかった。
宮下 敦巳; 大沼 敏治*; 酒井 高行*; 岩沢 美佐子*; 吉川 正人; 叶野 琢磨; 曽根田 直樹*
no journal, ,
SiC半導体デバイスは、耐放射線に優れ、高電圧・高温での動作が可能なことから、従来のSiやGaAs半導体デバイスでは動作が困難な、原子炉や宇宙環境等,極限環境下で用いられる素子として期待されている。しかしSiCデバイスの特性を左右する酸化膜界面には、Siデバイスでの酸化膜界面に比べて界面欠陥が多く存在しているため、SiCデバイス特性は理論的に予測される値よりも遥かに低い値しか実現できていない。また、物理的測定手法から推定される界面欠陥の原子構造から電気特性を推定することは困難である。そこで、第一原理分子動力学法を用いて計算機上に界面欠陥構造を生成し、エネルギー準位や荷電状態等の特性を算出することで、界面欠陥の物理構造とその電気特性との関連性を明らかにし、加えてSiC結晶表面の酸化膜成長メカニズムを明確にする。これによりSiCデバイスの電気特性を最大限に引き出せる物理的界面形成法の開発指針を得る。結晶SiO/SiC中規模モデルに対して、SiO側終端固定の条件において、加熱温度4000K,加熱時間3ps及び急冷速度-1000K/psの条件のもとで、界面ダングリングボンドのない急峻なアモルファスSiO/SiC界面構造を生成し、エネルギー準位や電荷分布等が導出できた。また、中規模モデルを用いて界面酸化反応の模擬計算を行い、酸素分子が連続して界面に達した場合に起こるSiO/SiC界面の酸化過程の第一原理分子動力学計算を世界で初めて成功させた。
/4H-SiC(0001)界面における熱酸化過程の第一原理分子動力学シミュレーション; 炭素クラスターの形成大沼 敏治*; 宮下 敦巳; 岩沢 美佐子*; 吉川 正人; 土田 秀一*
no journal, ,
平面波基底のPAW(Projector Augment Wave)法による第一原理分子動力学計算を行い熱酸化過程の動的シミュレーションを行った。計算は地球シミュレーターで行った。界面モデルはスラブモデルを用い、界面の初期構造は加熱及び急冷法により作成した。界面モデルは急峻かつダングリングボンドのない綺麗な界面に酸化過程のきっかけとして炭素空孔を導入したものを用いた。酸化過程のシミュレーションは酸素分子を一つずつSiO層に追加することにより行った。酸素分子はSiO層中及びSiC界面のSi原子と反応し解離した。SiC界面のSi原子が酸化されることによりSi原子とC原子との結合が切れてCダングリングボンドが生成される。Cダングリングボンドの生成がきっかけとなり炭素クラスターが生成されるのが観察された。CO分子はCクラスタと酸素分子が反応することにより生成された。
/SiC界面の生成; 第一原理分子動力学計算宮下 敦巳; 大沼 敏治*; 岩沢 美佐子*; 土田 秀一*; 吉川 正人
no journal, ,
SiCデバイス絶縁膜を模擬するために、加熱・急冷計算によりアモルファスSiO/SiC界面構造を計算機上に構築し電子構造を決定した。計算は地球シミュレータ上で第一原理分子動力学計算コード(VASP)を用いて行った。444原子を含む構造モデルを用いた加熱・急冷計算において、初期構造では界面Si原子に存在していたダングリングボンドが、室温まで冷却された最終構造では完全に消滅した急峻界面が生成されていた。この時の加熱条件は4000K/3ps、冷却条件は-1000K/psである。Si-Oの最近接原子間距離は0.165nmであり、 水晶の0.161nmとほぼ等しい。Si-Siの最近接原子間距離は0.315nmであり、これをSi-O-Siの結合角に換算すると145
水晶の0.161nmとほぼ等しい。Si-Siの最近接原子間距離は0.315nmであり、これをSi-O-Siの結合角に換算すると145 となりシリカガラスでのSi-O-Siの結合角(145
となりシリカガラスでのSi-O-Siの結合角(145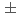 10)と合致する。さらに、O-Oの再近接原子間距離は0.266nm であり、これをO-Si-Oの結合角に換算すると107となり四面体配位での結合角109.5とほぼ等しい。短距離秩序が結晶の値にほぼ回復したのに対して長距離秩序は回復しておらず、これにより加熱・急冷計算でアモルファスSiO層が生成できたことが確認できた。
10)と合致する。さらに、O-Oの再近接原子間距離は0.266nm であり、これをO-Si-Oの結合角に換算すると107となり四面体配位での結合角109.5とほぼ等しい。短距離秩序が結晶の値にほぼ回復したのに対して長距離秩序は回復しておらず、これにより加熱・急冷計算でアモルファスSiO層が生成できたことが確認できた。
とUOにおける原子間力ポテンシャルの特性の研究中村 仁一; 金田 保則*; Chen, Y.*; Geng, H.*; 岩沢 美佐子*; 大沼 敏治*; 木下 幹康
no journal, ,
照射環境下で核燃料内に発生するさまざまな欠陥構造の理論的解明には、分子動力学法を用いた解析が有効である。特に面状欠陥の発生に着目し、この計算による再現を目標とした、UO並びに模擬物質としてのCeOの原子間ポテンシャルの開発・特性解析の現状について述べる。
宮下 敦巳; 大沼 敏治*; 酒井 高行*; 岩沢 美佐子*; 叶野 琢磨; 曽根田 直樹*; 吉川 正人
no journal, ,
SiC半導体は極限環境における次世代のMOS型デバイスとして有望であると期待されているが、界面においては界面欠陥密度が高くチャンネル移動度が低いことが知られている。この界面欠陥の性質を理解し、その形成過程を解明するには、計算機上で実デバイスに近い界面を生成し、さらに熱酸化過程のメカニズムを明らかにすることが重要である。1000原子規模の界面モデルを用いたアモルファス界面モデル生成における加熱・急冷計算で、室温までの冷却速度を-2000K/psから-500K/psまで遅くすることにより、界面における欠陥構造が緩和されることがわかった。また、Si面よりC面の酸化速度が速くなる原因が酸化反応の理論計算から確かめられた。
界面構造の第一原理計算による生成,3宮下 敦巳; 大沼 敏治*; 岩沢 美佐子*; 土田 秀一*; 吉川 正人
no journal, ,
第一原理分子動力学計算コードを用いた加熱・急冷計算によってアモルファス界面構造を構築し、電子状態が界面電気特性に与える影響を理論的側面から追求した。1017原子界面構造モデルに対して、冷却速度を従来用いていた-2000K/psから、-1000K/ps, -500K/psと変化させた所、室温冷却後の全エネルギーは-2000K/psの時を基準にして、それぞれ-7.2eV, -13.9eV低くなり安定化した。また、SiO層中でのSi-O-Si結合角も、135から、それぞれ137, 140と広くなり、よりシリカガラスでのSi-O-Siの結合角(14510)に近くなった。これらの結果から、冷却速度が遅い方が安定で実デバイスに近い界面構造を得られることがわかった。
 -
-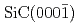 C面における酸化過程の第一原理分子動力学計算
C面における酸化過程の第一原理分子動力学計算大沼 敏治*; 宮下 敦巳; 岩沢 美佐子*; 吉川 正人; 土田 秀一*
no journal, ,
ワイドギャップ半導体であるSiCは次世代のMOS型パワーデバイスとして有望である。しかし 界面においては界面トラップ密度が高いことやMOSデバイスのチャンネル移動度が低いことが知られている。また界面においては界面遷移層が存在することが知られており、その存在が界面トラップ密度の高さやチャンネル移動度の低下に影響を及ぼしている可能性がある。界面遷移層の形成過程を解明するには、界面の熱酸化過程のメカニズムを明らかにすることが重要である。本報告では、C面における熱酸化過程の動的シミュレーションについて報告する。酸化過程のシミュレーションは酸素分子を一つずつSiO層に追加することにより行った。C面の熱酸化過程において酸素分子はSiO層中及びSiC界面のSi原子と反応し解離した。SiC界面のSi原子が酸化されることによりSi原子とC原子との結合が切れてCダングリングボンドが生成される。界面の炭素原子が酸化されてCO及びCO分子,C-C-O複合体が生成した。生成したCO
界面においては界面トラップ密度が高いことやMOSデバイスのチャンネル移動度が低いことが知られている。また界面においては界面遷移層が存在することが知られており、その存在が界面トラップ密度の高さやチャンネル移動度の低下に影響を及ぼしている可能性がある。界面遷移層の形成過程を解明するには、界面の熱酸化過程のメカニズムを明らかにすることが重要である。本報告では、C面における熱酸化過程の動的シミュレーションについて報告する。酸化過程のシミュレーションは酸素分子を一つずつSiO層に追加することにより行った。C面の熱酸化過程において酸素分子はSiO層中及びSiC界面のSi原子と反応し解離した。SiC界面のSi原子が酸化されることによりSi原子とC原子との結合が切れてCダングリングボンドが生成される。界面の炭素原子が酸化されてCO及びCO分子,C-C-O複合体が生成した。生成したCO 分子はCダングリングボンドと反応しない場合にSiO層を拡散する。酸化が進むと炭素の5員環構造などが生じるのが観察された。
分子はCダングリングボンドと反応しない場合にSiO層を拡散する。酸化が進むと炭素の5員環構造などが生じるのが観察された。
木下 幹康; 安永 和史*; 園田 健*; 岩瀬 彰宏*; 石川 法人; 左高 正雄; 安田 和弘*; 松村 晶*; Geng, H. Y.*; 一宮 尚志*; et al.
no journal, ,
新クロスオーバ研究で得られた成果概要を示す。高燃焼度燃料におけるセラミックス燃料の細粒化現象についてそのメカニズム解明とシミュレーション手法の開発をすすめている。主要な強調点は以下の2点である。(1)酸化ウランの模擬資料であるセリア(CeO)を用いて、イオンインプランターによる燃焼度の模擬、タンデム加速器による核分裂照射の模擬、の2つを組合せることによって細粒化の基礎過程である亜結晶の生成を実現した。(2)実燃料で観察される、上記細粒化の発生直前の事象である、酸素の再配置現象について、計算科学において検討をすすめた。過剰な酸素が存在する場合について(過化学量論組成について)第一原理計算では形成エネルギーの計算により秩序配列を再現することができた。加速分子動力学法では、過剰な酸素原子がクラスターをつくり、常温を含む低温でも高速に拡散運動することを初めて発見した。
; Cub-octahedoral clustersGeng, H. Y.*; Chen, Y.*; 岩沢 美佐子*; 大沼 敏治*; 木下 幹康
no journal, ,
The stability and energetics of oxygen defect clusters in UO+x are studied by first-principles method. The results show that the well known Willis-type cluster is actually unstable and the most possible clustering mode is in cub-octahedral geometry, whose competition with point defects determines the materials' low energy behavior.
Chen, Y.*; 金田 保則*; Geng, H. Y.*; Shang, J. C.*; 岩沢 美佐子*; 大沼 敏治*; 園田 健*; 一宮 尚志*; 鈴土 知明; 板倉 充洋; et al.
no journal, ,
新クロスオーバー研究の研究成果について、計算科学によるシミュレーション研究(第一原理電子論,分子動力学,統計力学・熱力学,メゾスコピック)の成果並びに、加速器によるシミュレーション実験の成果内容も含めてその概要を示す。また、今後の発展の方向と課題について述べる。
/UOにおける原子間ポテンシャルの特性金田 保則*; Geng, H. Y.*; Chen, Y.*; 岩沢 美佐子*; 大沼 敏治*; 木下 幹康
no journal, ,
高燃焼度状態でのUOに見られる細粒化現象には、核分裂照射による転位の生成・運動・Xe原子の存在などさまざまな要因が絡んでいると考えられている。また、実験的参照物質としてのCeOに対する模擬照射実験では、格子間酸素原子集合体なども観測されている。今回は、この格子間酸素原子クラスター核nIoの熱的安定性に対する計算・解析を行い、CeO/UOにおいて、そのイオン結合性とバルクの構造を再現する原子間ポテンシャルを用いることにより、(111)面上格子間酸素クラスター核が、2000K程度の高温でも比較的安定に存在しうることを計算により示す。
界面の大規模数値解析宮下 敦巳; 大沼 敏治*; 岩沢 美佐子*; 土田 秀一*; 吉川 正人
no journal, ,
Siに比べ優れた物理特性を持つSiCを基板に用いたSiC-MOSFETは、従来のSi半導体デバイスでは動作が困難な極限環境下で用いられる素子として期待されている。しかしSiCと酸化膜の界面には、SiC-MOSFETのデバイス特性を劣化させる原因である界面欠陥が多く存在しており、欠陥の原子構造とデバイス特性との関連性を追求することは非常に重要な問題となっている。本研究では実際のデバイスの酸化膜界面を模擬するようなアモルファス界面を計算機上に構築して、その原子構造を解析することで、電子状態が界面電気特性に与える影響を追求している。1017原子界面構造モデル(加熱・急冷時693原子)に対して、4000K 3psの固定終端加熱,終端解放後、3500K2psの継続加熱、-2000K/psで室温までの急冷を行った。生成したSiO層の動径分布関数を評価した所、Si-O結合距離は0.165nm、Si-O-Si結合角は135deg、O-Si-O結合角は109degと得られ、かつ、局所密度分布からも良好なアモルファス特性を示していることが確かめられた。
3psの固定終端加熱,終端解放後、3500K2psの継続加熱、-2000K/psで室温までの急冷を行った。生成したSiO層の動径分布関数を評価した所、Si-O結合距離は0.165nm、Si-O-Si結合角は135deg、O-Si-O結合角は109degと得られ、かつ、局所密度分布からも良好なアモルファス特性を示していることが確かめられた。
/SiC interface defect structure generated with first-principles molecular dynamics simulation宮下 敦巳; 大沼 敏治*; 岩沢 美佐子*; 土田 秀一*; 吉川 正人
no journal, ,
Siに比べ優れた物理特性を持つSiCを用いた半導体デバイスは、従来のSiやGaAs半導体デバイスでは動作が困難な極限環境下で用いられる素子として期待されているが、SiCと酸化膜の界面にはSiCデバイス特性を劣化させる界面欠陥が多く存在しており、その欠陥構造とデバイス特性との関連性を追求することは非常に重要である。本研究では実際の界面を模擬した原子構造モデルを計算機上に生成し、電子状態が界面電気特性に与える影響を理論的側面から追求している。表面固定の1017原子界面構造モデルに対して、4000K3psでの加熱,固定解放後3500K2psでの継続加熱,-2000K/psの速度で室温までの急冷を行った結果、生成されたモデルはSi-O結合距離が0.165nm, O-Si-O結合角が109, Si-O-Si結合角が135となり、SiO層が良好なアモルファス構造になったことが確かめられた。室温冷却後の界面構造では、従来モデルで想定されていた、Si原子が界面にある2つないしは3つのSi-O結合をまとめる構造以外にも、Si原子が一つのSi-O結合にのみ接続する構造,Si-Si結合を含む構造,界面Siのダングリングボンドが認められた。
