


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
山崎 智*; 寺田 透*; 河野 秀俊; 清水 謙多郎*; 皿井 明倫*
Nucleic Acids Research, 40(17), p.e129_1 - e129_7, 2012/09
被引用回数:20 パーセンタイル:44.47(Biochemistry & Molecular Biology)Proteins recognize a specific DNA sequence not only through direct contact (direct readout) with base pairs but also through sequence-dependent conformation and/or flexibility of DNA (indirect readout). However, it is difficult to assess the contribution of indirect readout to the sequence specificity. What is needed is a straightforward method for quantifying its contributions to specificity. Using Bayesian statistics, we derived the probability of a particular sequence for a given DNA structure from the trajectories of molecular dynamics (MD) simulations of DNAs containing all possible tetramer sequences. Then, we quantified the specificity of indirect readout based on the information entropy associated with the probability. We tested this method with known structures of protein-DNA complexes. This method enabled us to correctly predict those regions where experiments suggested the involvement of indirect readout. The results also indicated new regions where the indirect readout mechanism makes major contributions to the recognition. The present method can be used to estimate the contribution of indirect readout without approximations to the distributions in the conformational ensembles of DNA, and would serve as a powerful tool to study the mechanism of protein-DNA recognition.
山崎 智*; 寺田 透*; 清水 謙多郎*; 河野 秀俊; 皿井 明倫*
Nucleic Acids Research, 37(20), p.e135_1 - e135_9, 2009/09
被引用回数:7 パーセンタイル:14.52(Biochemistry & Molecular Biology)Proteins recognize DNA sequences by two different mechanisms. The first is direct readout, in which recognition is mediated by direct interactions between the protein and the DNA bases. The second is indirect readout, which is caused by the dependence of conformation and the deformability of the DNA structure on the sequence. Various energy functions have been proposed to evaluate the contribution of indirect readout to the free-energy changes in complex formations. We developed a new generalized energy function to estimate the dependence of the deformability of DNA on the sequence. This function was derived from molecular dynamics (MD) simulations previously conducted on B-DNA dodecamers, each of which had one possible tetramer sequence embedded at its center. By taking the logarithm of the probability distribution function (PDF) for the base-step parameters of the central base-pair step of the tetramer, its ability to distinguish the native sequence from random ones was superior to that with the previous method that approximated the energy function in harmonic form. From a comparison of the energy profiles calculated with these two methods, we found that the harmonic approximation caused significant errors in the conformational energies of the tetramers that adopted multiple stable conformations.
皿井 明倫*; 河野 秀俊
実験医学, 26(7), p.1099 - 1105, 2008/04
転写因子などのDNA結合タンパク質は、遺伝子の制御に重要な役割を果たしている。タンパク質は、通常複数のDNA配列を認識し複数の遺伝子の発現を制御しているが、タンパク質があるDNAを特異的に認識する機構についてはまだよくわかっていない。したがって、タンパク質とDNAの特異的結合を正確に予測することは容易ではない。また、構造ゲノミクスプロジェクトによって機能未知のタンパク質の立体構造が次々と明らかにされている現在、それらのタンパク質がDNAに結合するかどうか、また結合するならば、どこの部位を使って結合するかといった分子機能を予測することも重要になってきている。本稿ではこれまでに開発されたDNA結合タンパク質の予測方法,結合部位の予測方法,タンパク質の結合塩基配列の予測方法、特にわれわれが開発してきた構造情報を用いる方法について解説し、今後の展望について述べる。
藤井 聡*; 河野 秀俊; 竹中 繁織*; 郷 信広; 皿井 明倫*
Nucleic Acids Research, 35(18), p.6063 - 6074, 2007/09
被引用回数:106 パーセンタイル:87.05(Biochemistry & Molecular Biology)タンパク質がDNAを認識するには、DNAとの水素結合や静電相互作用による直接的な認識とDNAの構造特性に由来した間接的な認識がある。われわれは、後者の間接的な認識を定量化するために、DNAの構造特性を分子動力学計算によって調べた。結果、これまで2塩基対で特徴づけられていた構造は、その特性を記述するのに不十分であること、塩基対の配列依存性が2つ先の塩基対までかなり影響することなどを示した。さらに、間接認識ポテンシャルを作成し、それが細胞内で見られるDNAのヌクレオソーム構造形領域を予測できることを示した。
皿井 明倫*; 河野 秀俊
実験医学, 25(10), p.77 - 84, 2007/06
真核生物の転写制御系では多くの転写因子の組合せがいろいろな遺伝子を複雑に制御している。そこでは、素子である転写因子の分子レベルですでに高度な協同性が働いていると思われる。本稿では、転写因子によるDNA配列の認識メカニズムと、協同性の役割について構造的な観点から解説する。さまざまな転写因子によるDNAの複合体構造の解析から、協同性が、転写因子とDNAの構造変化と密接に結びついており、認識の特異性を高めることを示す。
米谷 佳晃*; 河野 秀俊; 藤井 聡*; 皿井 明倫*; 郷 信広
Molecular Simulation, 33(1-2), p.103 - 107, 2007/01
被引用回数:5 パーセンタイル:16.09(Chemistry, Physical)5'AATT3'と5'TTAA3'の2種類の塩基配列のDNAについて分子動力学シミュレーションを行い、構造変化と水和の関係を調べた。シミュレーションから、5'AATT3'では、DNAは構造変化しにくく、水和水は構造化しやすいが、5'TTAA3'では、DNAは構造変化しやすく、水和水は構造化しやすいことが明らかになった。この結果に基づいてDNAの構造変化と水和の関係について議論した。
Ahmad, S.*; 河野 秀俊; Ara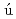 zo-Bravo, M. J.*; 皿井 明倫*
zo-Bravo, M. J.*; 皿井 明倫*
Nucleic Acids Research, 34(Suppl.2), p.W124 - W127, 2006/07
被引用回数:31 パーセンタイル:48.89(Biochemistry & Molecular Biology)転写因子などのDNA結合蛋白質はDNA配列に特異的に結合する。DNA結合蛋白質のDNA配列の認識は、おもに静電相互作用,水素結合,ファンデルワールス相互作用などの塩基とアミノ酸残基の直接的な相互作用によって行われていると考えられてきた。しかし、多くの蛋白質-DNA複合体構造が明らかになるにつれ、直接的な相互作用の数が少ないこと,直接に蛋白質と相互作用をしていない塩基配列を変えても結合強度が変わることなどが明らかになり、直接的でない認識、つまり、間接認識の重要性も認知されるようになってきた。間接認識とは、配列に依存したDNAの特異構造や曲がりやすさなどの構造情報や物性情報を通してDNA配列を蛋白質が間接的に認識することをさす。したがって、蛋白質とDNAの認識機構を解明するには、この両者の寄与を明らかにする必要がある。しかしながら、直接認識と間接認識の寄与を定量化することが難しいため、その寄与バランスはよくわかっていない。われわれは、両者の寄与を定量化する方法を開発し、研究者がさまざまな蛋白質-DNA複合体についてその寄与バランスを知ることができるように、寄与バランスを計算できるツールをインターネット上に公開した。
Gromiha, M. M.*; Siebers, J. G.*; Selvaraj, S.*; 河野 秀俊; 皿井 明倫*
Gene, 364, p.108 - 113, 2005/12
被引用回数:28 パーセンタイル:45.90(Genetics & Heredity)蛋白質のDNA認識は蛋白質とDNAの分子間相互作用とDNA自身の分子内相互作用に分けて考えることができるが、その両者の寄与バランスはよくわかっていない。われわれは、最近その定量化方法を提案している。その方法を用いて多くの蛋白質とDNAの複合体を評価し、蛋白質とDNAの相互作用から計算される配列特異性への寄与とDNA構造の変形がもたらす配列特異性への寄与バランスを調べた。その結果、転写因子は幅広いバランスを持つが、酵素は分子間特異性が強いことがわかった。
Arazo-Bravo, M. J.*; 藤井 聡*; 河野 秀俊; Ahmad, S.*; 皿井 明倫*
Journal of the American Chemical Society, 127(46), p.16074 - 16089, 2005/11
被引用回数:46 パーセンタイル:72.44(Chemistry, Multidisciplinary)すべてのテトラマー配列(136通り)を12merのDNA配列の真中にもつDNAの分子動力学計算を行い、DNA構造,ダイナミクスの配列依存性を解析した。その結果、ピリミジン-プリンステップを持つ配列は非常に前後の配列の影響を受けやすいことがわかった。また、分子動力学計算から得られた構造アンサンブルから統計ポテンシャルを計算し、簡便にDNAの構造エネルギーを評価できるようにした。
皿井 明倫*; 河野 秀俊
Annual Review of Biophysics and Biomolecular Structure, 34, p.379 - 398, 2005/06
被引用回数:155 パーセンタイル:78.93(Biochemistry & Molecular Biology)蛋白質とDNAの相互作用について、構造生物学的観点からレビューする。蛋白質のDNA認識は、塩基とアミノ酸の直接的な認識とDNA自体の構造変形を認識する間接認識とに分けて考えることができる。多くの蛋白質とDNAの複合体構造を定量的に評価した結果、蛋白質のDNA認識における配列特異性は、この2つのほぼ同等な寄与により達成されていた。
皿井 明倫*; Siebers, J. G.*; Selvaraj, S.*; Gromiha, M. M.*; 河野 秀俊
Journal of Bioinformatics and Computational Biology, 3(1), p.169 - 183, 2005/02
構造バイオインフォマティクスと計算生物学を融合して蛋白質のDNA認識機構について調べた。既に多くの蛋白質とDNAの複合体が解かれており、その立体構造をもとに蛋白質-DNA相互作用の統計ポテンシャルを作成し、蛋白質によるDNA認識の特異性を定量化した。また、計算機シミュレーションにより、DNA3塩基対とアミノ酸残基1つからなる小さな系で、塩基対まわりの自由エネルギー面を計算した。この自由エネルギー面は、実際に観測される塩基のまわりのアミノ酸分布をよく再現しており、自由エネルギー面から蛋白質のDNA認識を考察することができた。
米谷 佳晃; 藤井 聡*; 皿井 明倫*; 河野 秀俊; 郷 信広
no journal, ,
DNAの構造揺らぎは塩基配列に依存し、その配列による違いが蛋白質との結合親和性を左右する。したがって、DNAの構造揺らぎの塩基配列依存性は、蛋白質との相互作用を考えるうえで重要な性質である。一方、DNAの水和構造も塩基配列に依存する。例えば、AATTなど特定の配列で、副溝に沿って水分子が連なる秩序構造が出現することがわかっている。本研究では、全4塩基配列パターンについて分子動力学計算を行い、DNAの構造ゆらぎと副溝に見られる水和パターンの関係を系統的に調べた。その結果、すべての塩基対ステップはおもに4通りのグループに分類され、その分類に従って、構造ゆらぎと水和パターンの間に成り立つ関係を説明できることがわかった。さらに、DNAの構造ゆらぎと水和パターンの間に相関が現われた原因を調べるために、水分子のDNA水和サイト滞在時間などを計算し、DNAの塩基対ステップと水和水の運動が原子レベルで呼応している様子を捉えた。
藤井 聡*; 河野 秀俊; 皿井 明倫*
no journal, ,
The conformational properties of DNA play important roles in various biological processes. For example, interactions of proteins with DNA sequences are known to be affected by the conformational properties of DNA. It has also been known that the transitions among various conformational states are induced by the change in environments: Variations in ionic strength and identity, sequence, water activity, and ligand/protein binding all can modulate DNA structure. In this study, we analyzed how the sequence and the environments such as water activity influence the DNA conformations. We have carried out a series of molecular dynamics (MD) simulations to explore the conformational preferences and dynamical behavior of DNA duplex. We prepared the initial structure models with two different sequences in two kinds of conformations; d(CGCGTATACGCG)2 and d(CGCGTATACGCG)2 in Band A-forms. The MD simulations were carried out in four different environments: in only water solvent and in 75, 85 and 95 % (v/v) ethanol / water mixture solvent. We analyzed the differences of the DNA backbone conformation, the base-pair step parameter and the localization of water molecules in the groove on each structure. The variability of the conformation was different depending on the solvation condition. The DNA conformation seemed to be influenced by the localization of the water molecules in the groove.
藤井 聡*; 河野 秀俊; 竹中 繁織*; 郷 信広; 皿井 明倫*
no journal, ,
DNAが関係する多くの生体機能には、DNAの配列依存的な構造や物性が関係している。例えば、小分子や蛋白質とDNAとが相互作用する際、DNAの構造特性が影響することが知られている。そのような認識において、DNAの外観、つまりDNAのリン酸骨格のコンフォーメーションも重要な役割を果たしていると考えられる。そこで、このようなDNAの配列依存的な構造特性を調べるためにFreeのDNAに対して分子動力学計算を行い、リン酸骨格の構造に着目して解析を行った。ユニークなテトラマー136種類(AATT, AAAC, CGATなど)を含む12塩基対の二本鎖DNA d(CGCGWXYZCGCG)2 (WXYZ:テトラマー)について、それぞれ10ns分子動力学計算を行い配列ごとのtorsion angleの分布を比較した。特徴的であったのは、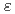 /
/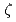 における2個のコンフォーメーションの安定状態(BI/BII)であった。このBII状態を取る頻度には配列依存性が現れていた。また、蛋白質-DNA複合体の結晶構造を調べたところ、BII構造が特徴的な部位に存在していた。したがって、蛋白質-DNA認識においてこのようなリン酸骨格の構造特性が重要であると考えられる。
における2個のコンフォーメーションの安定状態(BI/BII)であった。このBII状態を取る頻度には配列依存性が現れていた。また、蛋白質-DNA複合体の結晶構造を調べたところ、BII構造が特徴的な部位に存在していた。したがって、蛋白質-DNA認識においてこのようなリン酸骨格の構造特性が重要であると考えられる。
米谷 佳晃*; 藤井 聡*; 皿井 明倫*; 河野 秀俊; 郷 信広
no journal, ,
DNAの柔らかさは塩基配列に依存し、その配列による違いが蛋白質との複合体形成の親和性を大きく左右することがある。そのため、DNAの柔らかさの塩基配列依存性は、蛋白質との相互作用を考えるうえで重要な性質である。一方、DNAの水和構造も塩基配列に依存することが知られている。例えば、配列5'AATT3'の場合には、副溝に沿って水分子が連なる秩序構造が形成されるが、5'TTAA3'の場合には形成されにくいことが、X線結晶構造解析から示されている。このような水和秩序構造の発見により、それがB型DNAの構造安定性に影響しているのではないかと考えられるようになったが、さまざまな配列における水和構造とDNAの柔らかさの関係についてはよくわかっていない。本研究では、両者の関係を明らかにするために、DNAの柔らかさと水和構造をさまざまな配列について系統的に調査した。DNAの柔らかさについては、既に4塩基配列の全配列パターン(136通り)について分子動力学計算を行い、その配列依存性を導いている。今回は、さらに136通りの配列について水和構造を解析した。導かれた水和構造をDNAの柔らかさと比較し、DNAの柔らかさに対する水和構造の変化とその配列による違いを示した。水和構造とDNAの柔らかさが互いにどのように影響しあっているのかを議論する。
米谷 佳晃*; 藤井 聡*; 皿井 明倫*; 河野 秀俊; 郷 信広
no journal, ,
DNAの柔らかさは塩基配列に依存し、その配列による違いが蛋白質との複合体形成の親和性を大きく左右することがある。そのため、DNAの柔らかさの塩基配列依存性は、蛋白質との相互作用を考えるうえで重要な性質である。一方、DNAの水和構造も塩基配列に依存することが知られている。例えば、配列5'AATT3'の場合には、副溝に沿って水分子が連なった秩序構造(スパイン)が形成されるが、5'TTAA3'の場合には形成されにくいことが、これまでのX線結晶構造解析から示されている。このような水和秩序構造の発見により、それがB型DNAの構造安定性に影響しているのではないかと考えられるようになったが、さまざまな配列に対して、水和構造とDNAの柔らかさがどのように関係しているかよくわかっていない。本研究では、両者の関係を明らかにするために、DNAの柔らかさと水和構造をさまざまな配列について系統的に調査した。DNAの柔らかさについては、すでに4塩基配列の全配列パターン(136通り)について分子動力学計算を行い、その配列依存性を導いている。今回は、さらに136通りの配列パターンについて水和構造を解析した。導かれた水和構造をDNAの柔らかさと比較し、DNAの柔らかさに対する水和構造の変化、そしてその配列による違いを示した。水和構造とDNAの柔らかさが互いにどのように影響しあっているのかを議論する。
米谷 佳晃*; 藤井 聡*; 皿井 明倫*; 河野 秀俊; 郷 信広
no journal, ,
DNAの柔らかさは塩基配列に依存し、その配列による違いが蛋白質との複合体形成の親和性を大きく左右することがある。そのため、DNAの柔らかさの塩基配列依存性は、蛋白質との相互作用を考えるうえで重要な性質である。一方、DNAの水和構造も塩基配列に依存することが知られている。例えば、配列5'AATT3'の場合には、副溝に沿って水分子が連なった秩序構造(スパイン)が形成されるが、5'TTAA3'の場合には形成されにくいことが、これまでのX線結晶構造解析から示されている。このような水和秩序構造の発見により、それがB型DNAの構造安定性に影響しているのではないかと考えられるようになったが、さまざまな配列に対して、水和構造とDNAの柔らかさがどのように関係しているかよくわかっていない。本研究では、両者の関係を明らかにするために、DNAの柔らかさと水和構造をさまざまな配列について系統的に調査した。DNAの柔らかさについては、すでに4塩基配列の全配列パターン(136通り)について分子動力学計算を行い、その配列依存性を導いている。今回は、さらに136通りの配列パターンについて水和構造を解析した。導かれた水和構造をDNAの柔らかさと比較し、DNAの柔らかさに対する水和構造の変化、そしてその配列による違いを示した。水和構造とDNAの柔らかさが互いにどのように影響しあっているのかを議論する。
米谷 佳晃*; 藤井 聡*; 皿井 明倫*; 河野 秀俊; 郷 信広
no journal, ,
DNAの柔らかさは塩基配列に依存し、その配列による違いが蛋白質との複合体形成の親和性を大きく左右することがある。そのため、DNAの柔らかさの塩基配列依存性は、蛋白質との相互作用を考えるうえで重要である。一方、DNAの水和構造も塩基配列に依存することが知られている。例えば、AATTなどの配列では、副溝に沿って水分子が連なった秩序構造が形成されることが、これまでのX線結晶構造解析から示されている。このような水和秩序構造の発見により、それがDNAの構造安定性に影響しているのではないかと考えられるようになったが、さまざまな配列に対して水和パターンとDNAの柔らかさがどのように関係しているかよくわかっていない。本研究では、4塩基配列の全配列パターンについて分子動力学計算を行い、DNAの柔らかさと水和パターンの間に成り立つ関係を系統的に調査した。その結果、DNAの柔らかさ水和パターンの間に明確な相関が現れることを明らかにした。さらに、このような相関が見られる原因を調査し、ブリッジ形成がDNAの柔らかさを決定づけるというよりむしろ、DNAの柔らかさが水和パターンを決定づけていることを示唆する結果を得た。
米谷 佳晃; 藤井 聡*; 皿井 明倫*; 河野 秀俊; 郷 信広
no journal, ,
Sequence dependence of DNA conformational deformability is one of important factors in the sequence-specific recognition by regulatory DNA-binding proteins. Intrinsic mechanical properties of DNA will make a dominant contribution, but the other effect of hydration cannot be neglected. A well known water spine is composed of highly ordered water molecules in the minor groove of specific DNA sequences. Emergence of such a hydration pattern depends on the sequence, raising the following question. How is the hydration related with the conformational properties of DNA? To address this question, we analyzed the deformability and hydration of various DNA sequences using the ensembles of DNA conformations generated by molecular dynamics simulations. The result showed a correlation between the DNA deformability and the hydration pattern. It was suggested that the obtained relation is associated with the coupled motions of DNA base-pair steps and hydration water.
藤井 聡*; 河野 秀俊; 郷 信広*; 皿井 明倫*
no journal, ,
Sequence-dependent conformational properties of DNA play important roles in various biological processes. For example, interactions of proteins with DNA sequences are known to be affected by the conformational properties of DNA. Proteins such as TATA-box binding protein and catalytic protein DNaseI induce the DNA conformation to bent form and A-form at recognition sites, respectively. It has been known that these conformational transitions are induced by the environment: variations in ionic strength and identity, sequence, water activity, and ligand/protein binding all can modulate DNA structure. In this study, we analyzed how the sequence and water activity influence the DNA conformations using molecular dynamics simulations.
