


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
本庄 栄二郎; 玉田 太郎; 安達 基泰; 黒木 良太; Meher, A.*; Blaber, M.*
Journal of Synchrotron Radiation, 15(3), p.285 - 287, 2008/05
被引用回数:5 パーセンタイル:29.06(Instruments & Instrumentation)われわれは薄板状にしか結晶化しないhaFGFの分子間接触部位を改善し結晶成長を制御することを試みている。haFGFのX線結晶構造から結晶学的に対称な位置関係にあるhaFGF分子の分子間接触部位にはGlu81の側鎖が近接して存在しており、この電荷の反発が分子間相互作用を乱している可能性が考えられた。このGlu81の側鎖が分子間相互作用に関与しているかどうかを調べるため、われわれはGlu81をAla, Val, Leu, Ser及びThrに置換したhaFGFの変異体を作成した。このGlu81の側鎖が分子間相互作用に関与しているかどうかを調べるため、われわれはGlu81をAla, Val, Leu, Ser及びThrに置換したhaFGFの変異体を作成した。それぞれの変異体について蟻酸を用いて結晶化を行ったところ、Ala, Val及びLeu変異体は野生型同様薄板状の結晶成長が見られたが、Ser及びThr変異体の結晶は野生型よりもより厚みが増していた。これらの変異体について1.5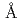 分解能のX線構造を解いたところ分子間接触部位に存在するSerの水酸基が沈殿剤である蟻酸を介して水素結合を形成していることがわかった。このような分子間接触部位改変の試みは中性子結晶構造解析のための大型結晶育成に寄与すると考えられる。
分解能のX線構造を解いたところ分子間接触部位に存在するSerの水酸基が沈殿剤である蟻酸を介して水素結合を形成していることがわかった。このような分子間接触部位改変の試みは中性子結晶構造解析のための大型結晶育成に寄与すると考えられる。
新井 栄揮; 玉田 太郎; 前田 宜丈*; 黒木 良太
no journal, ,
マウス抗体TN1は、巨核球系細胞の増殖・分化及び血小板産生を促進するサイトカインであるヒト・トロンボポエチン(hTPO)を認識する。われわれは、TN1抗体によるhTPO中和活性の発現機構の解明のために、TN1由来Fab単体のX線結晶構造を2.0オングストローム分解能で決定し、TN1由来Fab-hTPO複合体中のFabの構造と比較した。その結果、抗原結合時と比較して、非結合時はFabのFv領域・C1領域の相対位置が大きくずれることが明らかになった。抗原結合・非結合時のFab全体のC を比較すると、RMSDは2.4オングストロームである。一方、TN1抗体によるhTPO認識は、超可変領域(CDR)の主鎖構造がほとんど変化せず、ごくわずかに生じた側鎖レベルでのInduced-fitに基づくことが明らかになった。この結果は、TPO結合の際、TN1抗体のCDRが大きな構造変化を伴わないことを示唆する。
を比較すると、RMSDは2.4オングストロームである。一方、TN1抗体によるhTPO認識は、超可変領域(CDR)の主鎖構造がほとんど変化せず、ごくわずかに生じた側鎖レベルでのInduced-fitに基づくことが明らかになった。この結果は、TPO結合の際、TN1抗体のCDRが大きな構造変化を伴わないことを示唆する。
安達 基泰; 玉田 太郎; 栗原 和男; 大賀 拓史*; 倉光 成紀*; 黒木 良太
no journal, ,
ADPリボースピロフォスファターゼ(ADPRase)は、ADPリボースをリボース5'リン酸とAMPに加水分解する酵素である。本研究では、中性子結晶構造解析によって、ADPRaseの触媒反応に関与する水分子の役割や解離性アミノ酸のイオン化状態を明らかにし、触媒機構を詳細に解明することを目的としている。大腸菌を用いて組換え型ADPRaseを調製後、マクロシーディング法によって3.2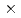 2.81.5(13mm
2.81.5(13mm )の結晶を取得した。得られた結晶を20日間重水素化溶液中でソーキングを行った後、JRR-3の回折計BIX-3を用いて回折データを測定した。予備的解析実験の結果、露光時間240分で2.1オングストロームの回折データが得られた。さらに、ADPRaseの完全重水素化の取り組みについても報告する。
)の結晶を取得した。得られた結晶を20日間重水素化溶液中でソーキングを行った後、JRR-3の回折計BIX-3を用いて回折データを測定した。予備的解析実験の結果、露光時間240分で2.1オングストロームの回折データが得られた。さらに、ADPRaseの完全重水素化の取り組みについても報告する。
玉田 太郎; 本庄 栄二郎; 前田 宜丈*; 岡本 智之*; 石橋 松二郎*; 徳永 正雄*; 黒木 良太
no journal, ,
ヒト顆粒球コロニー刺激因子(GCSF)とそのヒト受容体(GCSF-R)中のリガンド結合領域との複合体の活性構造を2.8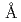 分解能で決定した。GCSF/GCSF-R複合体中の構成比は2:2で、GCSF-R中のIg-likeドメインとGCSFがたすき掛けすることにより二量体化していた。この結合様式はヒトGCSFとマウスGCSF-R(CRH)ドメイン複合体中の様式とは全く異なっており、インターロイキン6とその受容体であるgp130との活性複合体中で確認された様式と類似していた。このIg-likeドメインを介したGCSF-Rの二量体化はこれまでに報告されている熱力学的及び変異体解析の結果と相関性があった。
分解能で決定した。GCSF/GCSF-R複合体中の構成比は2:2で、GCSF-R中のIg-likeドメインとGCSFがたすき掛けすることにより二量体化していた。この結合様式はヒトGCSFとマウスGCSF-R(CRH)ドメイン複合体中の様式とは全く異なっており、インターロイキン6とその受容体であるgp130との活性複合体中で確認された様式と類似していた。このIg-likeドメインを介したGCSF-Rの二量体化はこれまでに報告されている熱力学的及び変異体解析の結果と相関性があった。
正山 祥生; 玉田 太郎; 竹内 彩子*; 田浦 太志*; 正山 征洋*; 黒木 良太; 森元 聡*
no journal, ,
THCA synthase is the enzyme that catalyzes oxidative cyclization of cannabigerolic acid into THCA, the precursor of Delta1-tetrahydrocannabinol. In order to investigate the structure-function relationship of THCA synthase, this enzyme was overproduced in insect cells, purified and finally crystallized in 0.1 M HEPES buffer pH 7.5 containing 1.4 M sodium citrate. A single crystal suitable for X-ray diffraction measurement was obtained in 0.09 M HEPES buffer pH 7.5 containing 1.26 M sodium citrate. The crystal diffracted to 2.8 resolution at beamline BL41XU, SPring-8. The crystal belonged to the primitive cubic space group P432, with unit-cell parameters a=b=c=178.2 . R value of the structure model was 19.6%. Active site of THCA synthase was investigated by mutation analysis using the structural information from X-ray crystallography. It was found that Tyr484 was identified to be one of the important residues in activity of THCA synthase.
栗原 和男; 玉田 太郎; 大原 高志; 黒木 良太
no journal, ,
Hydrogen atoms and hydration water molecules in proteins are indispensable components on many naturally-occurring processes. Neutron crystallography is used to analyze the role of hydrogen in the structure and function of bio-macromolecules. There are two neutron diffractometers for biological macromolecules (BIX-3 and BIX-4) installed at the Japan Atomic Energy Agency (JAEA). They were constructed by Dr. Niimura and his group, and since 2004 the present authors have maintained and developed them. Data collection for a total of 14 proteins and DNA oligomers has been successful using BIX-3 and BIX-4. Data for 8 samples of them have been collected since 2004. From these experiments using BIX-3 and BIX-4, we have summarized an empirical relation between crystal volume, unit cell volume, and diffraction resolution. This provides prior estimate of the quality of neutron measurement on BIX-3 and 4 for given crystal size and cell constants.
山口 繁生*; 上久保 裕生*; 栗原 和男; 清水 哲哉*; 山崎 洋一*; 黒木 良太; 新村 信雄*; 片岡 幹雄*
no journal, ,
Hydrogen atoms play important roles for many biochemical reactions. In the case of photoactive yellow protein (PYP), protonation/deprotonation reactions on amino acid residues and the following rearrangements in the hydrogen bond network are proposed to be closely related with the conformational changes. To understand the molecular mechanism, it is essential to determine the positions of hydrogen atoms. Neutron crystallographic and high resolution X-ray crystallographic studies are well known to be useful for observation of hydrogen atoms. We could obtain a fairly large and high quality crystal of PYP (2.89 0.85 0.79 mm) which gave X-ray diffraction spots up to 0.7. The high-resolution structural analysis clarified previously unknown hydrogen bonding network related with the tertiary structural change. To confirm the hydrogen positions, we are also carrying out neutron diffraction experiments with the obtained crystal.
大原 高志; 海野 久雄; 玉田 太郎; 黒木 良太
no journal, ,
蛋白質水和水は蛋白質分子及び隣接する水和水と水素結合ネットワークを形成し、プロトンの授受や運動の制御など蛋白質分子の機能発現に重要な役割を果たしていると考えられている。われわれは蛋白質の中性子構造解析結果を収録した「生体水素水和水データベース(HHDB)」を用い、蛋白質水和水と蛋白質分子中のカルボニル基、及び蛋白質水和水同士の水素結合ネットワークについて水素原子位置も含めた統計的解析を行った。その結果、O-H...O水素結合角が通常とは異なる統計的分布を示すことが明らかとなった。一般的に水素結合におけるX-H...Yの角度は180 近傍が安定とされ、有機低分子化合物や蛋白質分子内の水素結合では160
近傍が安定とされ、有機低分子化合物や蛋白質分子内の水素結合では160 170付近に数多く分布している。これに対し、本研究における解析では13590のO-H...O角度を持つ水素結合が多数分布していた。この原因について解析を進めたところ、蛋白質水和水が形成する水素結合ネットワーク中において、準安定な水素結合様式とされる"Bifurcated"及び"Inversed"水素結合が幾つも存在することが明らかとなった。
170付近に数多く分布している。これに対し、本研究における解析では13590のO-H...O角度を持つ水素結合が多数分布していた。この原因について解析を進めたところ、蛋白質水和水が形成する水素結合ネットワーク中において、準安定な水素結合様式とされる"Bifurcated"及び"Inversed"水素結合が幾つも存在することが明らかとなった。
茶竹 俊行*; 栗原 和男; 黒木 良太; 森本 幸生*
no journal, ,
Proteinase K (PK) has highly activity in various environments. The aim of our study is to carry out neutron crystallographic analysis of PK for revealing detailed mechanisms of reaction and a resistance to severe environments. As for PK, it is hardly to get large crystals suitable for neutron diffraction study. Moreover, many NO ions locate on the surface of this protein, and some amino acids seemed to be distorted. At first, we found a new crystallization condition. PK crystals have so excellent quality that 1.05 X-ray diffraction data could be collected using a synchrotron radiation. Next, crystallizations were carried out in the deuterated solution. D
ions locate on the surface of this protein, and some amino acids seemed to be distorted. At first, we found a new crystallization condition. PK crystals have so excellent quality that 1.05 X-ray diffraction data could be collected using a synchrotron radiation. Next, crystallizations were carried out in the deuterated solution. D O solution had a small influence on the crystallization, and this crystallization condition was adequate for neutron experiment. Still photographs of deuterated crystals were collected with 12 hours exposure time using BIX-3 neutron diffractometer at JRR-3. Several diffraction spots could be identified.
O solution had a small influence on the crystallization, and this crystallization condition was adequate for neutron experiment. Still photographs of deuterated crystals were collected with 12 hours exposure time using BIX-3 neutron diffractometer at JRR-3. Several diffraction spots could be identified.
