


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
塩谷 光平; 新山 友暁*; 下川 智嗣*
Materials Transactions, 66(6), p.704 - 711, 2025/04
被引用回数:0 パーセンタイル:0.00(Materials Science, Multidisciplinary)ハイエントロピー合金(HEA)は、ほぼ等量ずつの5種類以上の元素により構成される多元系合金である。本論文では、HEAにおける粒界移動の分子動力学シミュレーションを実施し、HEAの各構成元素の濃度が粒界移動挙動に及ぼす影響を系統的に調査した。解析の結果、HEAの粒界移動に必要な駆動力は、粒界の移動が間欠的になるか、あるいは粒界移動速度が低下する条件において最大になることがわかった。また、最大の駆動力は粒界への元素の偏析度合いが最大になるときに達成された。さらに、粒界偏析の度合いはHEAの構成元素組成によってさまざまに制御可能であり、粒界偏析は粒界の移動に必要な駆動力や粒界移動速度といった粒界の移動挙動に強く影響を与えることがわかった。本研究は、HEAの元素組成が粒界移動挙動を決定する上で重要な役割を果たすことを示し、得られた結果は優れた機械的特性を有するHEAの設計に貢献するものである。
小林 恵太; 奥村 雅彦; 中村 博樹; 板倉 充洋; 町田 昌彦; 浦田 新吾*; 鈴谷 賢太郎
Scientific Reports (Internet), 13, p.18721_1 - 18721_12, 2023/11
被引用回数:15 パーセンタイル:75.53(Multidisciplinary Sciences)非晶質物質の構造因子に現れる鋭い第一ピークはFSDPと呼ばれ、非晶質物質中の中距離秩序構造を反映したものであると考えられているが、その構造的起源に関しては現在まで議論が続いている。今回、第一原理計算と同等の精度を持つ機械学習分子動力学を用いることにより、高密度シリカガラスにおけるFSDPの構造起源を解析した。まず、機械学習分子動力学を用いることにより、高密度シリカガラスの様々な実験データの再現に成功した。また、高密度シリカガラスにおけるFSDPの発達(減少)は、ガラス構造の圧縮に伴う、Si-O共有結合ネットワーク中のリング構造の変形挙動によって特徴付けられることを明らかにした。
小林 恵太; 中村 博樹; 板倉 充洋; 町田 昌彦; 奥村 雅彦
まてりあ, 62(3), p.175 - 181, 2023/03
核燃料の研究開発において、原子炉運転時からシビアアクシデント時の融点付近に至る温度領域まで、核燃料物質の高温物性を把握することが必須となるが、その取扱いの困難さから、実験研究を行うことは容易ではない。一方、シミュレーション研究は安全に実施可能であるが、高温物性評価のために必要である、高精度な大規模構造の長時間シミュレーションは、従来のシミュレーション手法では、実施が難しかった。我々は、最近開発された機械学習技術を応用して高精度な大規模構造の長時間シミュレーションが実施可能な「機械学習分子動力学法」を用いて、酸化トリウムの高温熱物性評価に成功した。本稿は、機械学習分子動力学法と我々の研究成果について概説する。
小林 恵太; 奥村 雅彦; 中村 博樹; 板倉 充洋; 町田 昌彦; Cooper, M. W. D.*
Scientific Reports (Internet), 12(1), p.9808_1 - 9808_11, 2022/06
被引用回数:16 パーセンタイル:69.43(Multidisciplinary Sciences)核燃料の一つであるトリウム酸化物に対し、機械学習分子動力学法を用い、その高温物性を調査した。様々な交換汎関数により第一原理計算を実施し、ニューラルネットによりその結果を学習することにより機械学習力場を構築した。特にSCANと呼ばれる交換汎関数を用いた第一原理計算結果を学習することにより得られた機械学習力場は、ラムダ転移温度や融点を含め、比較可能な実験データの多くに対し高精度な結果を示した。
小林 恵太; 永井 佑紀; 板倉 充洋; 志賀 基之
Journal of Chemical Physics, 155(3), p.034106_1 - 034106_9, 2021/07
被引用回数:13 パーセンタイル:67.66(Chemistry, Physical)自己学習ハイブリッドモンテカルロ法は機械学習力場を利用することにより、第一原理計算の高速化を可能にする手法である。今回、自己学習ハイブリッドモンテカルロ法をNPTアンサンブルに適用し、液体シリカの解析を行った。本論文では自己学習ハイブリッドモンテカルロ法を用いることにより、厳密に第一原理計算の精度を保ちながら、高速に液体シリカの配置空間のサンプリングが可能であることを示した。また、液体シリカの構造因子の計算を行い実験データとの比較を行ったところ、両者はよい一致を見せた。
小林 恵太; 中村 博樹; 山口 瑛子; 板倉 充洋; 町田 昌彦; 奥村 雅彦
Computational Materials Science, 188, p.110173_1 - 110173_14, 2021/02
被引用回数:25 パーセンタイル:75.68(Materials Science, Multidisciplinary)セメント水和物(セメントペースト)は建築材はもとより、放射性セシウムの閉じ込め材料として利用される。本論文はセメント水和物の代表的なモデル物質であるトバモライトの機械学習力場の構築を行ったものである。トバモライトに対し第一原理計算を実施し、様々な原子配置とそのポテンシャルデータを大量に生成し、ニューラルネットを用いた機械学習力場の学習を行った。構築した機械学習力場はトバモライトの弾性係数,振動状態密度をほぼ第一原理と同等の精度で計算可能であることを確かめた。また、機械学習分子動力学法を実行し、トバモライト細孔における水,イオンの輸送特性の解析を行った。
木本 和志*; 河村 雄行*; 牧野 仁史
Journal of Computer Chemistry, Japan, 19(2), p.46 - 49, 2020/00
本研究は、モンモリロナイトの締固めをシミュレートするための、2次元粗視化分子動力学法を提案するものである。この方法では、モンモリロナイト分子の単位構造と水和水を一つの粒子に粗視化して、変形するモンモリロナイト分子を1次元的に連結された粗視化粒子で表現する。粗視化粒子間に働く力には、分子内の結合力と分子間ファンデルワールス力を考慮し、前者は調和振動子として、後者はレナードジョーンズポテンシャルでモデル化する。このようなモデルを用いて締固めの数値シミュレーションを行った結果、大きく変形したモンモリロナイト分子が、締固めの結果4から6分子程度積層することが明らかとなった。また、シミュレーション結果からX線回折パターンを合成したところ、実験でも観測される回折ピークが得られることが明らかとなった。
町田 昌彦; 加藤 孝一郎*; 志賀 基之
Journal of Chemical Physics, 148(10), p.102324_1 - 102324_11, 2018/03
被引用回数:23 パーセンタイル:71.14(Chemistry, Physical)水,重水,トリチウム水の液体状態における構造を第一原理経路積分分子動力学法を用いて研究した。用いた計算手法は、電子と原子核のすべてを量子力学的に取り扱うものであり、現時点で最も正確な計算手法の一つである。シミュレーションした結果は、実験とおおよそ一致しており、核量子効果により、同位体間の構造上の違いが明らかにされた。特に特筆すべき結果は、水分子内のOH共有結合中のHが隣接水分子との水素結合上の中点まで移動できる一方、重水素,トリチウムの順に中点まで行ける確率が減少するという傾向を得たことである。しかしながら、実験との完全な一致は未だ得られておらず、密度汎関数法の改良が必須であることが示唆された。また、最近の研究成果である密度汎関数に対するvan der Waals補正は液体状態における同位体効果を研究する上で必須であることが分かった。
海老原 健一; 蕪木 英雄; 板倉 充洋
「鋼の機械的特性に及ぼす水素の効果とその評価」シンポジウム予稿集(USB Flash Drive), 6 Pages, 2014/09
水素脆化は鉄鋼材料の強度低下や破壊をもたらす要因の1つであり、その機構の解明が求められている。高強度鋼の遅れ破壊や溶接部の低温割れは、偏析水素による粒界強度低下が主な原因と考えており、その機構は応力腐食割れと同様と考えられる。粒界強度低下に基づく水素脆化モデルでは、水素による粒界強度低下を原子レベルスケールの計算で評価し、その情報を用い巨視的スケールの強度やき裂進展の評価がなされているが、両スケール間におけるスケールのモデル化についてはあまり明確でない。特に、微視き裂先端での応力集中について、き裂周囲を弾性体とするモデルがあるが、その妥当性も明確でない。本研究では、微視き裂を含みその周囲に転位がない系の引張によるき裂周囲の応力を分子動力学(MD)と連続体計算(FEM)の計算手法で評価し、その差異について考察した。その結果、1%以下の低いひずみでは、両手法による応力分布は同様の結果となったが、それ以上になるとき裂先端の応力集中部から両者の差が大きくなった。また、応力集中のモデル化については、き裂周辺を単なる弾性体とするだけではMDの結果を再現できなかった。
岡本 芳浩; Madden, P. A.*
Journal of Physics and Chemistry of Solids, 66(1), p.448 - 451, 2005/01
LaCl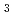 融体の局所構造は(LaCl
融体の局所構造は(LaCl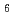 )
)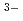 八面体配位によって特徴付けられている。これはX線回折やラマン散乱測定からの結論であるが、中性子回折や分子動力学計算では配位数はカチオンサイズによって変化し、比較的大きいLaClの場合は6より大きな値であることを示されている。本研究では、LaCl融体のX線回折を行い配位数について調べるとともに、中性子回折の結果と良好な一致をもたらす分子動力学計算との比較を行った。さらに、LaBr融体との構造比較を行い、アニオンサイズの違いに起因する以外、構造がほとんど同じであることをつきとめた。
八面体配位によって特徴付けられている。これはX線回折やラマン散乱測定からの結論であるが、中性子回折や分子動力学計算では配位数はカチオンサイズによって変化し、比較的大きいLaClの場合は6より大きな値であることを示されている。本研究では、LaCl融体のX線回折を行い配位数について調べるとともに、中性子回折の結果と良好な一致をもたらす分子動力学計算との比較を行った。さらに、LaBr融体との構造比較を行い、アニオンサイズの違いに起因する以外、構造がほとんど同じであることをつきとめた。
岡本 芳浩
Nuclear Instruments and Methods in Physics Research A, 526(3), p.572 - 583, 2004/07
被引用回数:32 パーセンタイル:86.38(Instruments & Instrumentation)溶融塩に代表される高温液体、つまり構造無秩序系のX線吸収スペクトル(XAFS)データを分子動力学(MD)計算から導き出す方法を考案した。従来は結晶系のXAFSシミュレーション用として用いられてきた多重散乱コードFEFFに、MD計算出力の位置データを入力データとして用いた。そのFEFF計算を数万回繰り返すことによって、MD計算の統計的積算が、XAFSのデバイワーラー因子に代表される揺らぎ成分に反映されるかどうかを調べた。実験で得た白金と幾つかのハロゲン化物溶融塩のデータが、この手法によって良好に再現できることを確認した。
志賀 基之; 高柳 敏幸
Chemical Physics Letters, 378(5-6), p.539 - 547, 2003/09
被引用回数:7 パーセンタイル:21.16(Chemistry, Physical)水ダイマーアニオンについての平衡構造を経路積分分子動力学法によって計算した。過剰電子と原子核の運動の両方を量子力学的に取り扱った。水-水のポテンシャルは半経験的な関数を、電子-水の擬ポテンシャルについては、最近開発された解析関数を用いた。電子が水ダイマーに付加すると、クラスターの構造が極めてフロッピーになることが計算によって示された。特に、ドナー交換,ドナー-アクセプター交換が量子力学的トンネル効果によって、容易に起こる事がわかった。このことは、核と電子の運動を分離するボルンオッペンハイマー近似が成り立たなくなっていることを示唆するものである。
岡本 芳浩; 赤堀 光雄; 伊藤 昭憲; 小川 徹
Journal of Nuclear Science and Technology, 39(Suppl.3), p.638 - 641, 2002/11
LiCl-KCl共晶塩中の、UCl融体の局所構造について、U原子L吸収端XAFS測定によって調べた。XAFS測定は、高エネルギー加速器研究機構の放射光実験施設BL27Bで実施した。ウラン水素化物の塩化によって調製した、UCl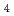 を亜鉛粉末で還元してUClを得た。カーブフィッティング解析の結果、最近接U
を亜鉛粉末で還元してUClを得た。カーブフィッティング解析の結果、最近接U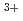 -Cl
-Cl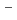 相関に関する構造情報を得た。MD計算とXAFSシミュレーションコードFEFF8の併用から、最近接U-Cl相関の相互作用について評価した。また、いくつかのウランハロゲン化物のXANES測定を行い、そのシフトから原子価について評価した。
相関に関する構造情報を得た。MD計算とXAFSシミュレーションコードFEFF8の併用から、最近接U-Cl相関の相互作用について評価した。また、いくつかのウランハロゲン化物のXANES測定を行い、そのシフトから原子価について評価した。
岡本 芳浩; 福島 和子*; 岩舘 泰彦*
Journal of Non-Crystalline Solids, 312(314), p.450 - 453, 2002/10
溶融臭化亜鉛の構造を、Zn及びBr双方のXAFSを測定することによって調べた。解析から、融体中で(ZnBr)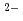 四面体錯体が存在し、Brイオンを共有することで複数の四面体がネットワーク構造を作り上げている可能性があることを示唆する結果が得られた。これは溶融ZnCl
四面体錯体が存在し、Brイオンを共有することで複数の四面体がネットワーク構造を作り上げている可能性があることを示唆する結果が得られた。これは溶融ZnCl の構造に良く似ている。得られたXAFSデータを、分子動力学計算とその出力を使用したFEFF計算から再現した。XAFSデータを良好に再現するMD計算では、四面体錯体の存在やネットワーク構造の存在を示す結果が得られた。
の構造に良く似ている。得られたXAFSデータを、分子動力学計算とその出力を使用したFEFF計算から再現した。XAFSデータを良好に再現するMD計算では、四面体錯体の存在やネットワーク構造の存在を示す結果が得られた。
清水 大志; 君塚 肇*; 蕪木 英雄; 荒川 忠一*
計算工学講演会論文集, 7(1), p.163 - 166, 2002/05
分子動力学法は現実の物質系をミクロなレベルからシミュレーションする手法であり、ニュートンの運動方程式を直接決定論的に数値積分することにより、系の静的な特性とともに動的な特性を求めることができる。分子動力学法シミュレーションにおいて最も計算時間を要する部分は一般的に原子間の相互作用の計算であり、大規模なシミュレーションを行なうには相互作用のカットオフにより計算コストを減らす高速化手法を用いることが必要である。また、今日では計算の高速化や大容量のメモリを確保するのに並列計算が有効である。われわれは、並列化やカットオフによる高速化などのシミュレーション手法を記述するプログラムの機能単位を切り分け、これらを再構成することにより並列分子動力学ステンシルを開発した。並列分子動力学ステンシルを利用すると、並列化手法及びカットオフによる高速化手法について意識することなくプログラミングを行なうことが可能である。本論文では、並列分子動力学ステンシルの設計及び性能について報告する。
清水 大志; 君塚 肇*; 蕪木 英雄; 荒川 忠一*
日本計算工学会論文集, 4, p.225 - 230, 2002/04
分子動力学法(MD)シミュレーションにおいて最も計算時間を要する部分は一般的に原子間の相互作用の計算である。各原子に作用する力を単純なアルゴリズムで計算するとペアを検索するコストが対象とする系の原子数の2乗に比例することから、短距離力の系について大規模なシミュレーションを行なう際は、粒子間に作用する力にカットオフを設定して計算コストを減らすカットオフ手法を用いることが必須となっている。また、計算の高速化や大容量のメモリを確保するのに並列計算が有効である。われわれは並列化などのシミュレーション手法の記述と系の性質の記述が分離した、見通しの良いプログラムを作成する枠組みとして並列分子動力学ステンシルを開発した。並列分子動力学ステンシルは、原子間相互作用を計算するプログラムを対象となる系の性質(モデル)の記述のみとなることを実現している。
板倉 憲一; 横川 三津夫; 清水 大志; 君塚 肇*; 蕪木 英雄
情報処理学会研究報告2001-HPC-88, p.67 - 72, 2001/10
地球シミュレータは、640の計算ノードを持ち理論ピーク性能は40Tflop/sである。プロセッサノードはピーク性能8Gflop/sのベクトルプロセッサ8個,16GBの共有メモリから構成される。本研究では地球シミュレータの計算ノードによる固体分子動力学法の計算プログラムのベクトル化と並列化を行い性能評価を行った。分子動力学法では、カットオフ半径内の粒子が互いに影響を与え、その粒子ペアを行列を用いて表現することができる。ベクトル化に際して、この行列表現にcompressed row formとjagged diagonalformを考える。jagged diagonal formはベクトル長がcompressedrow formよりも長くできるので、ベクトル化により適している。しかし、標準的な粒子対の情報からjagged diagonal formに変換するには時間がかかるため、全体の性能はより簡単なcompressedrow fromよりも低下した。compressed row formでは8CPUでの並列化により2.4から2.7倍のスピードアップとなった。
福本 一郎; 大村 悦二*
精密工学会誌, 67(6), p.916 - 921, 2001/06
高ピークパワーの超短パルスレーザーをターゲットに照射すると、照射領域が極短時間で蒸発し、熱伝導による影響の極めて小さい加工を行うことができる。そのため、精密・微細加工への応用が期待されている。超短パルスレーザーの加工分野への応用には、微視的アブレーションプロセスの解明が不可欠である。そこで、本論文では、金属ターゲットにガウシアンビームを照射した際に起こる微視的現象を、分子動力学法を用いてシミュレーションした。その結果、レーザー照射直後に表面下に強い衝撃波が発生し、表面上のレーザー照射部を起点として、円形に広がっていくことがわかった。また、この衝撃波の強度は、進行方向に対し一様ではなく、表面の垂線に対し15~40度の方向に強く現れることを示した。さらに、衝撃波が固液界面を通過する際、多数の転位が発生し、その一部は衝撃波とともに材料内に伝播することなどを明らかにした。
Bastug, T.*; Erkoc, S.*; 平田 勝; 館盛 勝一
Physica E, 8(3), p.223 - 229, 2000/09
被引用回数:5 パーセンタイル:32.03(Nanoscience & Nanotechnology)相対論密度汎関数法(RDFT)を用いて、ジルコニウム(Zr)2原子分子の構造最適化計算を行い、ポテンシャルエネルギー曲線を作成した。このポテンシャルを用いてZr3個から13個のクラスターの分子動力学(MD)シミュレーションを行ったのち、得られた最適化構造をもとにRDFTを用いて、Zr3個から7個のクラスターの電子状態を求めた。その結果、Zr3個から7個で構成されるクラスターは対称性が良く、8個から12個で構成されるクラスターは対称性が認められなかった。また、Zr13個で構成されるラスターは、Ih対称であり、最も対称性のよい最密構造をとることがわかった。
清水 大志; 君塚 肇*; 蕪木 英雄; Li, J.*; Yip, S.*
Proceedings of 4th International Conference on Supercomputing in Nuclear Applications (SNA 2000) (CD-ROM), 10 Pages, 2000/09
並列分子動力学(MD)について、さまざまな並列化手法やカットオフを用いた計算の効率化手法を組み合わせたシミュレーションをプログラムするのは簡単ではない。われわれは大規模な並列MDシミュレーションのためのプログラムの基本パターンである「並列分子動力学ステンシル」を開発した。本ステンシルは並列化手法や効率化手法を分離し、プログラム作成者がシミュレーション自体に専念できるように設計されている。プログラムの全体はC言語とMPIを用いるが、各MPI関数の呼び出しはプログラム作成者から隠蔽されている。本論文では並列MDステンシルを用い15~75Kの固体アルゴン(結晶状態及びアモルファス状態)に関して500~1,000,000原子を用いたシミュレーションを行い、その弾性について研究した。

