


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
 '(O'3)-type sodium cobalt oxide Na
'(O'3)-type sodium cobalt oxide Na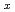 CoO
CoO (
(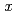
 0.78)
0.78)宮崎 譲*; 井川 直樹; 湯葢 邦夫*
Acta Crystallographica Section B; Structural Science, Crystal Engineering and Materials (Internet), 77(3), p.371 - 377, 2021/06
被引用回数:2 パーセンタイル:24.68(Chemistry, Multidisciplinary)Naイオン二次電池用正極材料としても期待される'(O'3)型層状コバルト酸ナトリウムNaCoO ( 0.78)について、中性子回折実験を実施し、Rietveld法によって結晶構造を解析した。その結晶構造は、(3+1)次元超空間群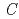 2 =
2 = 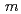 (
(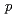 0
0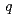 )00として表現される不整合変調を持つ構造であることを明らかにした。すなわち、Na原子の位置は、秩序的な配列をとるものの
)00として表現される不整合変調を持つ構造であることを明らかにした。すなわち、Na原子の位置は、秩序的な配列をとるものの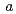 軸方向に最も大きく変調されている。このNaの疑似周期的な位置変調に伴って、CoOシート間のNaO
軸方向に最も大きく変調されている。このNaの疑似周期的な位置変調に伴って、CoOシート間のNaO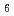 多面体は八面体型(O)からプリズム型(P)に変化する。Na原子が最も大きく変位している位置では、隣接するNa原子とほぼ接触した距離にあり、この位置ではNa原子の占有率が0となるNa欠損サイトが生成する。その結果、軸に沿って-O-O-O-P-
多面体は八面体型(O)からプリズム型(P)に変化する。Na原子が最も大きく変位している位置では、隣接するNa原子とほぼ接触した距離にあり、この位置ではNa原子の占有率が0となるNa欠損サイトが生成する。その結果、軸に沿って-O-O-O-P-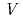
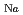 -P-パターンからなるNaO八面体連鎖が形成されることが分かった。
-P-パターンからなるNaO八面体連鎖が形成されることが分かった。
Sn and MgSi single crystals齋藤 亘*; 林 慶*; Huang, Z.*; 杉本 和哉*; 大山 研司*; 八方 直久*; 原田 正英; 及川 健一; 稲村 泰弘; 林 好一*; et al.
ACS Applied Energy Materials (Internet), 4(5), p.5123 - 5131, 2021/05
被引用回数:17 パーセンタイル:63.28(Chemistry, Physical)The development of thermoelectric (TE) materials, which can directly convert waste heat into electricity, is vital to reduce the use of fossil fuels. MgSn and MgSi are promising TE materials because of their superior TE performance. In this study, for future improvement of the TE performance, point defect engineering was applied to the MgSn and MgSi single crystals (SCs) via boron (B) doping. Their crystal structures were analyzed via white neutron holography and SC X-ray diffraction. Moreover, nanostructures and TE properties of the B-doped MgSn and MgSi SCs were investigated. The B-doping increased the chemical pressure on the MgSn and MgSi SCs, leading to induce vacancy defects as a point defect. No apparent change was observed in electronic transport, but thermal transport was significantly prevented. This study demonstrates that the vacancy defects can be controlled by the chemical pressure, and can aid in achieving a high TE performance for the MgSn and MgSi SCs.
Si single crystals" [AIP Advances 10, 035115 (2020)]林 慶*; 齋藤 亘*; 杉本 和哉*; 大山 研司*; 林 好一*; 八方 直久*; 原田 正英; 及川 健一; 稲村 泰弘; 宮崎 譲*
AIP Advances (Internet), 11(2), P. 029903_1, 2021/02
被引用回数:0 パーセンタイル:0.00(Nanoscience & Nanotechnology)MgSi is a potential thermoelectric (TE) material that can directly convert waste energy into electricity. In expectation of improving its TE performance by increasing electron carrier concentration, the element boron (B) is doped in MgSi single crystals (SCs). Their detailed crystal structures are definitely determined by using white neutron holography and single-crystal X-ray diffraction (SC-XRD) measurements. The white neutron holography measurement proves that the doped B atom successfully substitutes for the Mg site. The SC-XRD measurement confirms the B-doping site and also reveals the presence of the defect of Si vacancy (VSi) in the B-doped MgSi SCs. Regarding TE properties, the electrical conductivity, 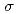 , and the Seebeck coefficient, S, decreases and increases, respectively, due to the decrease in the electron carrier concentration, contrary to the expectation. The power factor of the B-doped MgSi SCs evaluated from and S does not increase but rather decreases by the B-doping.
, and the Seebeck coefficient, S, decreases and increases, respectively, due to the decrease in the electron carrier concentration, contrary to the expectation. The power factor of the B-doped MgSi SCs evaluated from and S does not increase but rather decreases by the B-doping.
Si single crystals林 慶*; 齋藤 亘*; 杉本 和哉*; 大山 研司*; 林 好一*; 八方 直久*; 原田 正英; 及川 健一; 稲村 泰弘; 宮崎 譲*
AIP Advances (Internet), 10(3), p.035115_1 - 035115_7, 2020/03
被引用回数:19 パーセンタイル:69.37(Nanoscience & Nanotechnology)MgSi is a potential thermoelectric (TE) material that can directly convert waste energy into electricity. In expectation of improving its TE performance by increasing electron carrier concentration, the element boron (B) is doped in MgSi single crystals (SCs). Their detailed crystal structures are definitely determined by using white neutron holography and single-crystal X-ray diffraction (SC-XRD) measurements. The white neutron holography measurement proves that the doped B atom successfully substitutes for the Mg site. The SC-XRD measurement confirms the B-doping site and also reveals the presence of the defect of Si vacancy (VSi) in the B-doped MgSi SCs. Regarding TE properties, the electrical conductivity, , and the Seebeck coefficient, S, decreases and increases, respectively, due to the decrease in the electron carrier concentration, contrary to the expectation. The power factor of the B-doped MgSi SCs evaluated from and S does not increase but rather decreases by the B-doping.
elimination from 2,3-dimethylbutane cation: (CH )CHCH(CH)
)CHCH(CH)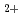
 (CH)C=C(CH)+H; An ab initio molecular orbital study
(CH)C=C(CH)+H; An ab initio molecular orbital study黒崎 譲*; 高柳 敏幸; 宮崎 哲郎*
Journal of Molecular Structure; THEOCHEM, 452, p.209 - 218, 1998/00
2,3-ジメチルブタンカチオン((CH)CHCH(CH),h-DMB )からのH脱離反応に対し、非経験的分子軌道計算を行った。構造最適化はUMP2/6-31G(d)レベルで行い、1点エネルギー計算をUMP3/6-31G(d)及びUMP4(SDTQ)/6-31G(d)レベルで行った。その結果、この反応は障壁が22-24kcal/molで26-29kcal/mol発熱的であることが予測された。非経験的分子軌道計算から得られたデータを用い、遷移状態理論に基づいて量子力学的(トンネル)効果を考慮した熱反応速度定数を求めると、h-DMBの反応の速度定数は77Kで約10
)からのH脱離反応に対し、非経験的分子軌道計算を行った。構造最適化はUMP2/6-31G(d)レベルで行い、1点エネルギー計算をUMP3/6-31G(d)及びUMP4(SDTQ)/6-31G(d)レベルで行った。その結果、この反応は障壁が22-24kcal/molで26-29kcal/mol発熱的であることが予測された。非経験的分子軌道計算から得られたデータを用い、遷移状態理論に基づいて量子力学的(トンネル)効果を考慮した熱反応速度定数を求めると、h-DMBの反応の速度定数は77Kで約10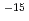 s
s と予測された。h-DMBにおいて、脱離するHをDで置換したカチオン(d-DMB)の反応の速度定数は77Kで約10
と予測された。h-DMBにおいて、脱離するHをDで置換したカチオン(d-DMB)の反応の速度定数は77Kで約10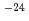 sと計算された。このことから、h-DMBからのH脱離反応にはトンネル効果が重要であることが示唆される。一方、h-DMBの反応速度定数に対する実測値は約12桁も大きい。これは量子化学計算のレベルがまだ低いことを示唆する。
sと計算された。このことから、h-DMBからのH脱離反応にはトンネル効果が重要であることが示唆される。一方、h-DMBの反応速度定数に対する実測値は約12桁も大きい。これは量子化学計算のレベルがまだ低いことを示唆する。
'-NaCoOの変調構造と輸送特性宮崎 譲*; 湯葢 邦夫*; 井川 直樹
no journal, ,
ナトリウムイオン電池の正極材料候補の一つである、層状コバルト酸化物'-NaCoOについて、中性子回折法によってその詳細な結晶構造を調べた。Rietveld解析の結果、本化合物のNa量は0.76(1)と求められた。基本構造は従来より提案されていた空間群2/で表され、その超空間群は2/(0)00であることが明らかになった。基本構造においては、原子はそれぞれNa(0, 1/2, 1/2), Co(0, 0, 0)、及びO(0.273, 0, 0.813)の座標を占め、対称性により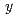 方向への変位変調は生じない。一方、Na原子の方向への変位変調が
方向への変位変調は生じない。一方、Na原子の方向への変位変調が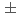 0.18と他の成分に比べて顕著に大きいことが明らかになった。
0.18と他の成分に比べて顕著に大きいことが明らかになった。
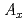 CoO(A=Na, Ca, Sr, Ba)における層間イオンの規則配列と熱電特性
CoO(A=Na, Ca, Sr, Ba)における層間イオンの規則配列と熱電特性五十嵐 大*; 宮崎 譲*; 湯葢 邦夫*; 井川 直樹; 梶谷 剛*
no journal, ,
Naサイトをアルカリ土類(Ca, Sr, Ba)で置換したCoO試料を合成し、層間イオンの価数と大きさがその規則配列及び熱電特性に及ぼす影響を調べた。中性子回折法によって、本試料はCoO層の周期に対して、A層の周期が不整合であり、(3+1)次元複合結晶であることが明らかになった。Ca系試料ではその分布に疎密が認められ、これがCo-O結合距離の変調を誘起させ、Coイオンの価数が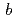 軸方向に周期的に電荷秩序が形成されていることが明らかになった。
軸方向に周期的に電荷秩序が形成されていることが明らかになった。
'-NaCoO ( 0.75)宮崎 譲*; 井川 直樹; 湯葢 邦夫*
no journal, ,
'(O1)型NaCoO ( 0.75)について、粉末中性子回折実験を行い、(3+1)次元の超空間群を仮定した解析によって、その変調構造を解析した。その結果、本構造は2/(0)00の超空間群として求めることができ、特に軸方向のNa原子の変調が重要であることがわかった。すなわち、Naはが約0.68の位置の場合-0.18までシフトするが、が0.32の位置では0.18へのシフトとなる。これらの場合、Na原子はその結晶サイト(0, 1/2, 1/2)から、近接原子に接触するほどの距離に相当する、およそ0.09nmも変位することになることが明らかになった。
'-NaCoOの変調構造宮崎 譲*; 井川 直樹; 湯葢 邦夫*; 梶谷 剛*
no journal, ,
これまで詳細な結晶構造が不明のままであった'-NaCoOについて、粉末中性子回折実験を行い、(3+1)次元の超空間群2/(0)00を仮定したRietveld解析によって、その変調構造を解明した。その結果、Na位置は、軸及び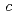 軸方向に大きく変調しており、周期的に欠損したサイトが存在すること、NaO多面体には八面体型とプリズム型の2種類があり、これらが軸方向に23個単位で交互に連結していること、CoO層も軸方向に大きく波を打っていることなどがわかった。
軸方向に大きく変調しており、周期的に欠損したサイトが存在すること、NaO多面体には八面体型とプリズム型の2種類があり、これらが軸方向に23個単位で交互に連結していること、CoO層も軸方向に大きく波を打っていることなどがわかった。
CoOの結晶構造と熱電特性五十嵐 大*; 宮崎 譲*; 湯葢 邦夫*; 井川 直樹; 梶谷 剛*
no journal, ,
層間カチオンによる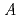 CoO(=Ca, Sr, Ba)の結晶構造変化について、粉末中性子回折及び電子回折によって調べた。粉末中性子回折パターンを比較するとBa
CoO(=Ca, Sr, Ba)の結晶構造変化について、粉末中性子回折及び電子回折によって調べた。粉末中性子回折パターンを比較するとBa CoOの002回折線は低角側に移動している。また、002と004回折線の間には層間Baの長周期構造に由来する回折線が観察された。本発表ではこれら層間の原子が作る規則配列と熱電性能の関係について議論する。
CoOの002回折線は低角側に移動している。また、002と004回折線の間には層間Baの長周期構造に由来する回折線が観察された。本発表ではこれら層間の原子が作る規則配列と熱電性能の関係について議論する。
