


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
黒崎 譲; 横山 啓一; 横山 淳
Journal of Molecular Structure; THEOCHEM, 913(1-3), p.38 - 42, 2009/11
We demonstrate, using optimal control theory, that perfect isotope-selective molecular vibrational excitations on the ground-state potential curve in multilevel systems can be completed in much shorter time scales than those in two-level systems. We consider a gaseous isotopic mixture of cesium iodide (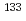 CsI and
CsI and 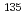 CsI) and, using two-level and multilevel systems, try to obtain electric fields that drive different isotopes into different vibrational levels. As a result, we find that in multilevel systems isotope-selective excitation processes can be controlled much faster, which we call multilevel effect. It is likely that this effect makes use of the large isotope shifts of higher vibrational levels than the lowest two.
CsI) and, using two-level and multilevel systems, try to obtain electric fields that drive different isotopes into different vibrational levels. As a result, we find that in multilevel systems isotope-selective excitation processes can be controlled much faster, which we call multilevel effect. It is likely that this effect makes use of the large isotope shifts of higher vibrational levels than the lowest two.
 SO
SO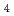
 (HO)
(HO)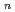 (
(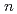 =1-6) on semiempirical PM6 potential surfaces
=1-6) on semiempirical PM6 potential surfaces柿崎 陽*; 茂木 春樹*; 吉川 武宏*; 高柳 敏幸*; 志賀 基之; 立川 仁典*
Journal of Molecular Structure; THEOCHEM, 901(1-3), p.1 - 8, 2009/05
半経験的PM6法を用いて、一連の硫酸クラスターを対象とした量子的な経路積分分子動力学シミュレーションを行った。クラスターサイズが大きくなるほど、イオン解離を起こしやすくなり、コンタクトイオンペアと呼ばれる構造が支配的となる。クラスター構造やプロトン移動過程において原子核の量子効果が重要であることを示す。
 O)
O)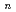 (=2-7) clusters on semiempirical PM6 potential energy surfaces
(=2-7) clusters on semiempirical PM6 potential energy surfaces高柳 敏幸*; 吉川 武宏*; 柿崎 陽*; 志賀 基之; 立川 仁典*
Journal of Molecular Structure; THEOCHEM, 869(1-3), p.29 - 36, 2008/11
半経験的分子軌道法であるPM6法に基づいた分子動力学法により、グリシン-水クラスターの熱力学的構造を調べた。クラスターサイズが大きくなるにつれグリシンが中性分子であるよりも双性イオンであるほうが安定になる傾向が見られた。また、中性分子と双性イオンの形での水和構造の違いについて議論する。
黒崎 譲
Journal of Molecular Structure; THEOCHEM, 850(1-3), p.9 - 16, 2008/02
被引用回数:12 パーセンタイル:29.10(Chemistry, Physical)最低励起三重項状態(T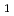 )のポテンシャルエネルギー面(PES)上でのアセトアルデヒドの光分解における水素原子生成チャンネル: CD
)のポテンシャルエネルギー面(PES)上でのアセトアルデヒドの光分解における水素原子生成チャンネル: CD CHO
CHO CDCHO+D(1); CDCHOCDCO+H(2)について、密度汎関数理論(DFT)に基づく直接分子動力学計算を実施した。DFT法はB3LYPで、基底関数はcc-pVDZを用いた。計算の結果、生成した水素原子の平均の運動エネルギーは反応1, 2でそれぞれ18.3, 16.6kcal mol
CDCHO+D(1); CDCHOCDCO+H(2)について、密度汎関数理論(DFT)に基づく直接分子動力学計算を実施した。DFT法はB3LYPで、基底関数はcc-pVDZを用いた。計算の結果、生成した水素原子の平均の運動エネルギーは反応1, 2でそれぞれ18.3, 16.6kcal mol であった。これらは、実験値[Kang et al., Chem. Phys. Lett. 434 (2007) 6]の半分から2/3の値であった。計算値が実験値に比べてかなり小さい理由は、反応1, 2の生成物側から見た障壁がB3LYP/cc-pVDZレベルでは小さく見積もられていることにある。しかしながら、ここでの計算結果は実験値と定性的には一致しており、これらの反応がTPES上で起こることを支持するものである。
であった。これらは、実験値[Kang et al., Chem. Phys. Lett. 434 (2007) 6]の半分から2/3の値であった。計算値が実験値に比べてかなり小さい理由は、反応1, 2の生成物側から見た障壁がB3LYP/cc-pVDZレベルでは小さく見積もられていることにある。しかしながら、ここでの計算結果は実験値と定性的には一致しており、これらの反応がTPES上で起こることを支持するものである。
藤本 浩文; Pinak, M.; 根本 俊行*; 坂本 清隆*; 山田 和幸*; 星 芳幸*; 久米 悦雄
Journal of Molecular Structure; THEOCHEM, 681(1-3), p.1 - 8, 2004/01
分子動力学シミュレーションプログラムAMBERと、AMBERの出力ファイルから動画ファイルを自動的に生成するために新たに開発したソフトウエアF-BMVSを組合せたシステムを用いて、複数箇所に酸化損傷を被ったDNA分子がどのように分子構造を変えるかを観察した。放射線や化学物質によって生じるこれらの損傷は、修復されなければ強力な突然変異原となり、細胞がガン化する原因となると考えられている。2種類の損傷(8-oxoG, AP部位)を持つDNA分子と損傷を持たないDNA分子に対し1ナノ秒のオーダーのシミュレーションを行い、F-BMVSを用いて可視化したところ、損傷構造に特異的な立体構造が観察された。これらの分子構造の差異は、損傷を修復する酵素が損傷部位と非損傷部位とを見分ける要因の1つになっているのではないかと考えられる。
Pinak, M.
Journal of Molecular Structure; THEOCHEM, 583(1-3), p.189 - 197, 2002/04
突然変異に関与する酸化DNA損傷8-オキソグアニン(8-oxoG)の局部的な構造とエネルギーへの影響を、分子動力学(MD)シミュレーションによって調べた。8-oxoGを15塩基対のB-DNAの中央に位置するグアニンと置換した系で、計算コードAMBER5.0を用いて2nsのMDシミュレーションを行った。損傷部分では、水素結合の崩壊が生じB-DNA構造が部分的にゆがむことが観測された。8-oxoGの向かいの(対をなす)シトシンに隣接するアデニンは、DNA二重らせんからはじき出されることがわかった。この構造の変化が、修復酵素が損傷DNAと結合する上で好都合であると推測される。また、DNA構造の不安定性は、8-oxoGを含むヌクレオチドと隣のヌクレオチドとの間の強い静電的な斥力により生じることことが示された。
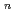 F
F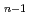 (n=2-5) clusters
(n=2-5) clusters羽毛田 直樹*; 横山 啓一; 田中 宏昌*; 工藤 博司*
Journal of Molecular Structure; THEOCHEM, 577(1), p.55 - 67, 2002/01
金属化合物やクラスターの電子状態を調べる目的で、リチウム原子が過剰に含まれるフッ化物クラスターLiF(n=2-5) の構造と電子構造を理論計算により求めた。その結果、過剰電子が分子全体に非局在化して分布することができるのはn=2のときだけであり、nが3以上では過剰電子が特定の部位に局在化して金属的電子状態が消失することがわかった。このことは、すでに研究されているLi(OH)クラスターに比べて、LiFクラスターではイオン結合性が強いことを示唆している。また、末端リチウム原子の数とイオン化エネルギーの相間がLi(OH)の場合と類似していることが示された。
H + Cl CHCl + Cl reaction; Ab initio molecular orbital study
+ Cl CHCl + Cl reaction; Ab initio molecular orbital study黒崎 譲
Journal of Molecular Structure; THEOCHEM, 545(1-3), p.225 - 232, 2001/07
反応CH + Cl CHCl + Clのポテンシャルエネルギー面を、CASSCF及びMRCIレベルでcc-pVTZ基底関数を用いて計算した。その結果、この反応はCHCl + Cl サイドから見て、CASSCFレベルでは小さな反応障壁をもつが、MRCIレベルでは反応障壁を持たないことが明らかとなった。すなわち、反応CHCl + Cl CH + Clは自発的な反応であることが予測された。MRCI計算の結果は、以前われわれが行ったPMP4(SDTQ)レベルでの計算結果 [J. Mol. Struct. (Theochem) 503 (2000) 231]を強く支持するものである。
NO( B)NO(
B)NO(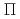 )+H reaction
)+H reaction黒崎 譲*; 高柳 敏幸
Journal of Molecular Structure; THEOCHEM, 507(1-3), p.119 - 126, 2000/07
反応HNO(2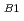 )NO()+Hの機構について非経験的分子軌道法を用いて理論的に研究した。基底状態と第一励起状態のポテンシャル面を適当な2つの内部座標の関数として、FOCI/cc-PVTZレベルで計算した。その結果、HNO(B)の分子面に垂直なCs面を保持した反応経路上に、conical intersectionが存在することが明らかとなった。同じ反応経路上には遷移状態が存在することも、既に明らかであったが、電子波動関数の対称性から、この遷移状態を経由する反応は対称禁制であることが予測されていた。しかし、今回の計算結果から、反応系はconical intersectionを回避する形で対称性をCsからCに落とすことにより禁制を解くということが明らかとなった。
)NO()+Hの機構について非経験的分子軌道法を用いて理論的に研究した。基底状態と第一励起状態のポテンシャル面を適当な2つの内部座標の関数として、FOCI/cc-PVTZレベルで計算した。その結果、HNO(B)の分子面に垂直なCs面を保持した反応経路上に、conical intersectionが存在することが明らかとなった。同じ反応経路上には遷移状態が存在することも、既に明らかであったが、電子波動関数の対称性から、この遷移状態を経由する反応は対称禁制であることが予測されていた。しかし、今回の計算結果から、反応系はconical intersectionを回避する形で対称性をCsからCに落とすことにより禁制を解くということが明らかとなった。
H+ClCHCl reaction黒崎 譲*
Journal of Molecular Structure; THEOCHEM, 503(3), p.231 - 240, 2000/05
本研究では、まず気相中における反応CH+ClCHClの機構について理論的に検討した。極限的反応座標(IRC)計算の結果、得られた反応物、遷移状態(TS)、生成物が1つの反応経路上にあることが確認された。反応の活性化エネルギーはPMP4及びB3LYPレベルで、それぞれ36.3,35.9kcal/molと計算された。次に、溶媒中における同反応の機構について検討した。その結果、極性溶媒中では気相中のようなTSが存在しないことが予測された。また、気相中では励起状態であったCHCl +Cl
+Cl が、極性溶媒中では基底状態となることが明らかとなった。誘電率80の極性溶媒中では、CHCl+Clのエネルギー値は反応物(CH+Cl)と比較して11.2kcal/molとなることがB3LYPレベルで計算された。このことは、この反応が気相中よりも極性溶媒中でより起こりやすいことを示唆している。
が、極性溶媒中では基底状態となることが明らかとなった。誘電率80の極性溶媒中では、CHCl+Clのエネルギー値は反応物(CH+Cl)と比較して11.2kcal/molとなることがB3LYPレベルで計算された。このことは、この反応が気相中よりも極性溶媒中でより起こりやすいことを示唆している。
Pinak, M.*
Journal of Molecular Structure; THEOCHEM, 499, p.57 - 70, 2000/03
DNA損傷のひとつであるチミンダイマー(TD)が修復酵素によって除去される過程は、酵素とDNAと複合体の形成に始まる。本研究では、酵素-DNA複合体形成のメカニズム解明のために、TDがあるDNAと、酵素の活性中心部位のみからなる系の500ピコ秒間の分子動力学シミュレーションを行った。TDは紫外線によって誘発されるDNA損傷、T4エンドヌクレアーゼVはTDに特異的な修復酵素である。これら2種類の分子を同一セル内に配置し、相対的な動きを計算した。特に、酵素-DNA複合体の形成における静電エネルギーの役割に重点を置いた。最初の100ピコ秒間に、酵素の一部は静電力とファンデルワールス力によってDNAの損傷部位に接近して結合し、500ピコ秒までの間安定な状態を維持した。計算の最初の段階で、酵素とTDの間の正の静電作用が働き、酵素はDNAに接近した。また、酵素とDNAの間に形成された水素結合が、複合体を安定に保つ役割を果たした。比較のために、損傷のないDNAについても同様の計算を行ったが、この場合には酵素とDNAの間には負の静電作用が働き、複合体は形成されなかった。
Pinak, M.*
Journal of Molecular Structure; THEOCHEM, 466, p.219 - 234, 1999/00
修復酵素によるDNA損傷の認識機構は、正確な修復のためには欠かせない。T4エンドヌクレアーゼVは、バクテリオファージT4由来のDNA修復酵素であり、チミンダイマー(TD)の修復過程の最初の段階を触媒する。TD部位を正確に認識する機構についての知見を得るために、損傷のないDNA、TDのあるDNA、T4エンドヌクレアーゼV、それぞれの分子について各々600ps間の分子動力学シミュレーション(MD)を行った。シミュレーション結果は、認識過程における静電作用の役割に着目して解析した。この結果、酵素のアミノ酸の静電エネルギーは、+15kcal/mol程度の正に荷電していることがわかった。TD部位の静電エネルギーは、-9kcal/mol程度の負に荷電しており、損傷のないDNAのチミン部位の中性の値とは異なった。TD部位の周囲の水との静電作用は、ほかのヌクレオチドの場合とは異なった。TDと損傷のないDNAのチミンとの違いは、静電エネルギーがTD部位を適切に認識するうえで重要な因子であることを示している。
D)+CH insertion reaction高柳 敏幸; 黒崎 譲*
Journal of Molecular Structure; THEOCHEM, 492, p.151 - 158, 1999/00
N(D)+CH反応について分子軌道法を直接用いた古典的トラジェクトリー計算を行った。反応の遷移状態から出発し、反応座標方向にエネルギーを与えて計算を行った。N(D)原子がCHのCH結合に挿入して生成するCHNH中間体ラジカルの寿命は非常に短いことがわかった。この結果は実験結果と定性的に一致する。このことからエネルギーはおもにCHとNHフラグメントの相対運動に分配されると予測することが可能である。
elimination from 2,3-dimethylbutane cation: (CH)CHCH(CH)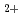 (CH)C=C(CH)+H; An ab initio molecular orbital study
(CH)C=C(CH)+H; An ab initio molecular orbital study黒崎 譲*; 高柳 敏幸; 宮崎 哲郎*
Journal of Molecular Structure; THEOCHEM, 452, p.209 - 218, 1998/00
2,3-ジメチルブタンカチオン((CH)CHCH(CH),h-DMB)からのH脱離反応に対し、非経験的分子軌道計算を行った。構造最適化はUMP2/6-31G(d)レベルで行い、1点エネルギー計算をUMP3/6-31G(d)及びUMP4(SDTQ)/6-31G(d)レベルで行った。その結果、この反応は障壁が22-24kcal/molで26-29kcal/mol発熱的であることが予測された。非経験的分子軌道計算から得られたデータを用い、遷移状態理論に基づいて量子力学的(トンネル)効果を考慮した熱反応速度定数を求めると、h-DMBの反応の速度定数は77Kで約10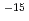 sと予測された。h-DMBにおいて、脱離するHをDで置換したカチオン(d-DMB)の反応の速度定数は77Kで約10
sと予測された。h-DMBにおいて、脱離するHをDで置換したカチオン(d-DMB)の反応の速度定数は77Kで約10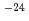 sと計算された。このことから、h-DMBからのH脱離反応にはトンネル効果が重要であることが示唆される。一方、h-DMBの反応速度定数に対する実測値は約12桁も大きい。これは量子化学計算のレベルがまだ低いことを示唆する。
sと計算された。このことから、h-DMBからのH脱離反応にはトンネル効果が重要であることが示唆される。一方、h-DMBの反応速度定数に対する実測値は約12桁も大きい。これは量子化学計算のレベルがまだ低いことを示唆する。
