


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
大沼 敏治*; 宮下 敦巳; 吉川 正人; 土田 秀一*; 岩沢 美佐子*
平成21年度先端研究施設共用促進事業「地球シミュレータ産業戦略利用プログラム」利用成果報告書, p.21 - 27, 2010/07
ワイドギャップ半導体である炭化珪素(SiC)は従来のシリコン(Si)半導体に比べて飛躍的な性能向上を実現するパワー半導体デバイスの材料として期待されている。また、SiC半導体デバイスは低損失の省エネデバイスとして開発が進められているとともに、Si半導体デバイスと同様に熱酸化により酸化絶縁膜を作製できるため、次世代のMOS型パワーデバイスとして有望である。しかし、従来のSiC MOS型パワーデバイスは、界面トラップの存在等によりチャネル移動度が理論的な予想値より遥かに小さく、優れた特性を発揮できていなかった。これらの特性を改善するためには、原子レベルで界面の構造と熱酸化の機構を明らかにすることが重要となる。SiCの熱酸化過程のシミュレーションにおいては、化学反応を伴うことと、界面においてさまざまな結合があることから、経験的なパラメータを一切用いない第一原理法が強力なツールとなるが、計算量が膨大なためこれまで行われてこなかった。地球シミュレータによる大規模な第一原理分子動力学計算によりSiCの熱酸化過程・アニーリング及び界面準位のシミュレーションが可能になったのでここに報告する。
 /SiC interface structure by the first-principles molecular dynamics simulation
/SiC interface structure by the first-principles molecular dynamics simulation宮下 敦巳; 大沼 敏治*; 岩沢 美佐子*; 土田 秀一*; 吉川 正人
Materials Science Forum, 556-557, p.521 - 524, 2007/00
SiCデバイスは宇宙や原子炉等の極限環境下で動作する素子として期待されている。しかしながら現状のSiCデバイスは理論的に予想されている性能を発揮しているとは言いがたい。その理由はSiCとその酸化膜であるSiOとの界面に存在する欠陥が素子の性能を低下させているからだと考えられる。実デバイスにある界面欠陥構造を計算機シミュレーションで再現しようとするなら、現実の界面にあるようなアモルファスSiO/SiCの構造を計算機上に再現することが非常に重要となってくる。われわれは444原子からなる結晶/結晶界面構造を計算機上に構築し、それに対して加熱・急冷計算を行うことでアモルファスSiO/SiC構造を生成した。加熱温度,加熱時間,急冷速度はそれぞれ4000K, 3ps, -1000K/psである。得られた界面構造のSiO領域はバルクのアモルファスSiO構造とよく適合し、界面におけるダングリングボンド欠陥も消滅していることが確かめられた。
/4H-SiC(0001) interface oxidation process; From first-principles大沼 敏治*; 宮下 敦巳; 岩沢 美佐子*; 吉川 正人; 土田 秀一*
Materials Science Forum, 556-557, p.615 - 620, 2007/00
平面波近似とスーパーセルモデルを用い、SiO/4H-SiC(0001)酸化過程の第一原理分子動力学計算による動的シミュレーションを行った。反応の初期構造の生成には加熱・急冷法を用いた。この初期構造は界面ダングリングボンドのないSiO/SiC構造である。酸化反応の引き金とするために、界面付近のSiC層に炭素空孔を導入した。酸化反応シミュレーションは界面付近の空隙に酸素分子を一つずつ置いて行くことによって行った。酸化反応シミュレーションは2500Kの下で行った。酸素分子は解離しSiO中のSi原子と結合を組み、また、界面付近にいるSiC層中のSi原子も酸化されSiO層を形成した。界面欠陥の候補の一つと考えられている炭素クラスタ構造が界面に形成され、さらに、酸素分子は炭素クラスターと反応しCO分子を形成した。
木下 幹康*; Geng, H. Y.*; Chen, Y.*; 金田 保則*; 岩沢 美佐子*; 大沼 敏治*; 園田 健*; 安永 和史*; 松村 晶*; 安田 和弘*; et al.
Proceedings of 2006 International Meeting on LWR Fuel Performance (TopFuel 2006) (CD-ROM), p.248 - 254, 2006/10
ニュー・クロスオーバー・プロジェクト(NXO)は、核燃料物質に対する核分裂放射線照射の影響を大学,国立・私立の研究所に渡る横断的な協力研究により検討している。加速器照射実験及び計算科学的手法を使用して、高燃焼度軽水炉燃料ペレットでリム構造を構成する機構を解明するためのシミュレーション研究が実施されている。加速器照射,高エネルギー電子線照射,核分裂エネルギー粒子線及び希ガス原子のイオン注入等が実施されている。照射試料としては、CeOを核燃料の模擬物質として使用した。最初の成果として、高エネルギー電子照射による平面構造形成、及び、高線量の高エネルギー粒子放射による結晶粒微細化に似た表面改質が観察された。第一原理及び分子動力学(MD)計算を用いた解析を実施し、UOとCeOの点欠陥形成エネルギー及び点欠陥を伴ったXe原子の相互作用についての情報が得られた。
/SiC interface宮下 敦巳; 吉川 正人; 叶野 琢磨; 大沼 敏治*; 酒井 高行*; 岩沢 美佐子*; 曽根田 直樹*
Annual Report of the Earth Simulator Center April 2004 - March 2005, p.287 - 291, 2005/12
Siに比べ優れた物理特性を持つSiCを用いた半導体デバイスは、従来のSiやGaAs半導体デバイスでは動作が困難な、原子炉や宇宙環境等、極限環境下で用いられる素子として期待されている。半導体素子界面では原子レベルの欠陥の荷電状態が電気特性を支配しているため、この界面構造を計算機上で模擬し、界面欠陥構造がどのようにデバイス特性に影響するのか導出するため、地球シミュレータを用いた第一原理分子動力学計算で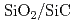 界面構造を構築し電子構造を決定する。400原子程度の中規模モデルを用いてアモルファス界面構造生成を行った。加熱温度は4000K、加熱時間は3ps、急冷速度は-1000K/ps、界面でのSiC可動層は4層とし、2200Kで
界面構造を構築し電子構造を決定する。400原子程度の中規模モデルを用いてアモルファス界面構造生成を行った。加熱温度は4000K、加熱時間は3ps、急冷速度は-1000K/ps、界面でのSiC可動層は4層とし、2200Kで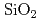 側終端固定層を開放し自由端とすることによって、層でのアモルファス化を促進させた。生成された界面はダングリングボンドが消滅しており、清浄界面に近い状態が再現されたが、バンドギャップ中には欠陥準位が存在するのが観察された。欠陥準位は界面に存在する酸素から生じており、結合に寄与できない局在した電子分布が準位の原因となっていることがわかった。
側終端固定層を開放し自由端とすることによって、層でのアモルファス化を促進させた。生成された界面はダングリングボンドが消滅しており、清浄界面に近い状態が再現されたが、バンドギャップ中には欠陥準位が存在するのが観察された。欠陥準位は界面に存在する酸素から生じており、結合に寄与できない局在した電子分布が準位の原因となっていることがわかった。
宮下 敦巳; 大沼 敏治*; 岩沢 美佐子*; 土田 秀一*; 吉川 正人
no journal, ,
第一原理分子動力学計算コードであるVASPコードを用いた加熱・急冷計算法による計算機シミュレーションによりアモルファスSiO/SiC界面構造の生成を行った。444原子界面構造モデルに対して、4000K, 3psで加熱融解、-1000K/psで室温までの急冷を行った。生成したSiO層の動径分布関数を評価したところ、全原子によるRDFでは長周期構造を反映した微細構造は認められず良好なアモルファス状態となっていた。部分RDFを評価したところ、Si-O結合距離は0.165nmであった。SiとSiの近接距離は約0.23nmに小さなピーク,0.315nmに大きなピークが認められる。0.23nmはSi-Si結合によるものでSiO中にSi-Si欠陥構造が存在することがわかる。0.315nmはSi-O-Si結合でのSi間距離に相当しSi-O-Si結合角は145 である。また、OとOの近接距離は0.266nmにピークを持ちO-Si-O結合角に換算すると107となった。これらの値はアモルファスSiOの条件に適合し、加熱・急冷計算によって良好なアモルファスSiO/SiC界面構造が生成されていることが確かめられた。
である。また、OとOの近接距離は0.266nmにピークを持ちO-Si-O結合角に換算すると107となった。これらの値はアモルファスSiOの条件に適合し、加熱・急冷計算によって良好なアモルファスSiO/SiC界面構造が生成されていることが確かめられた。
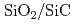 界面の生成
界面の生成宮下 敦巳; 大沼 敏治*; 吉川 正人; 岩沢 美佐子*; 中村 智宣*; 土田 秀一*
no journal, ,
Siに比べ優れた物理特性を持つSiCを用いた半導体デバイスは、従来のSiやGaAs半導体デバイスでは動作が困難な、原子炉や宇宙環境等、極限環境下で用いられる素子として期待されている。しかしながら、SiCの酸化膜とSiCの界面には界面欠陥が多く存在しており、この欠陥構造がSiCデバイスのチャネル移動度低下の原因となっている。半導体素子界面では原子構造が作り出す電子状態がその電気特性に影響を与えることから、界面欠陥構造とデバイス特性との関連性を追求することが重要である。本研究では、デバイス特性に影響を与える欠陥構造の解明を目標とし、欠陥を含んだ実際の界面に近い状態の原子構造を計算機上で模擬しエネルギー準位の計算を行っている。シミュレーション計算には地球シミュレータを用い、第一原理分子動力学計算による加熱・急冷計算によってアモルファス界面構造を構築し電子構造を決定した。シミュレーションには444原子の原子構造モデルを用い、加熱温度4000K、加熱時間3ps、急冷速度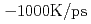 、終端以外の
、終端以外の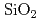 層の可動、界面のSiC可動層4層との条件を用いた。急冷時の2200Kにおいて終端固定層を開放し自由端とすることによって、層でのアモルファス化を促進させた。生成された界面は初期構造では界面Si原子に存在したダングリングボンドが消滅しており、清浄界面に近い状態が再現されていた。しかし、バンドギャップ中にはいまだ準位が存在し、欠陥準位は界面付近に存在する酸素原子の結合に寄与しない局在電荷分布に起因しているこがわかった。
層の可動、界面のSiC可動層4層との条件を用いた。急冷時の2200Kにおいて終端固定層を開放し自由端とすることによって、層でのアモルファス化を促進させた。生成された界面は初期構造では界面Si原子に存在したダングリングボンドが消滅しており、清浄界面に近い状態が再現されていた。しかし、バンドギャップ中にはいまだ準位が存在し、欠陥準位は界面付近に存在する酸素原子の結合に寄与しない局在電荷分布に起因しているこがわかった。
酸化膜のCV法による評価吉川 正人; 中村 智宣*; 宮下 敦巳; 大沼 敏治*; 土田 秀一*
no journal, ,
界面中間層のない急峻な界面形成を目指して、水素雰囲気下で表面をエッチングして平坦化させたエピ膜付4H-SiC基板表面に、シランと亜酸化窒素を用いてSiO膜を化学気層成長させた。電気的に良好な特性を有する堆積酸化膜形成条件をCV法で調べた結果、酸化膜堆積温度が850C以下で堆積後のアルゴンアニーリング温度が1100Cを超えると、界面の電気特性が良好になることがわかった。
/SiC界面構造の第一原理計算による生成宮下 敦巳; 大沼 敏治*; 岩沢 美佐子*; 吉川 正人; 土田 秀一*
no journal, ,
Siに比べ優れた物理特性を持つSiCを用いた半導体デバイスは、従来のSiやGaAs半導体デバイスでは動作が困難な、原子炉や宇宙環境等,極限環境下で用いられる素子として期待されている。しかしながら、SiCの酸化膜とSiCの界面には、SiCデバイスのチャネル移動度低下の原因となる界面欠陥が多く存在しており、その構造とデバイス特性との関連性を追求することは非常に重要である。本研究では実際の界面に近い状態の原子構造を計算機上で生成し、その電子状態が界面電気特性に与える影響を追求している。実際のデバイス絶縁膜を模擬するために、加熱・急冷計算によりアモルファスSiO/SiC界面構造を計算機上に構築し電子構造を決定した。計算には第一原理分子動力学計算コードであるVASPを用い、地球シミュレータ上で行った。444原子構造モデルを用いた加熱・急冷計算によって、初期構造では界面Si原子に存在していたダングリングボンドが完全に消滅した急峻界面が再現された。この時の条件は、加熱温度4000K,加熱時間3ps,急冷速度-1000K/psであり、さらに、急冷時の2200Kにおいて加熱中は固定してあったSiO終端を自由端とすることによって、SiO層での歪を低減させるとともにアモルファス化を促進させた。バンドギャップ中にはなお欠陥準位が存在したが、これはアモルファスSiO中の酸素のダングリングボンドに起因していることがわかった。
宮下 敦巳; 大沼 敏治*; 酒井 高行*; 岩沢 美佐子*; 吉川 正人; 叶野 琢磨; 曽根田 直樹*
no journal, ,
SiC半導体デバイスは、耐放射線に優れ、高電圧・高温での動作が可能なことから、従来のSiやGaAs半導体デバイスでは動作が困難な、原子炉や宇宙環境等,極限環境下で用いられる素子として期待されている。しかしSiCデバイスの特性を左右する酸化膜界面には、Siデバイスでの酸化膜界面に比べて界面欠陥が多く存在しているため、SiCデバイス特性は理論的に予測される値よりも遥かに低い値しか実現できていない。また、物理的測定手法から推定される界面欠陥の原子構造から電気特性を推定することは困難である。そこで、第一原理分子動力学法を用いて計算機上に界面欠陥構造を生成し、エネルギー準位や荷電状態等の特性を算出することで、界面欠陥の物理構造とその電気特性との関連性を明らかにし、加えてSiC結晶表面の酸化膜成長メカニズムを明確にする。これによりSiCデバイスの電気特性を最大限に引き出せる物理的界面形成法の開発指針を得る。結晶SiO/SiC中規模モデルに対して、SiO側終端固定の条件において、加熱温度4000K,加熱時間3ps及び急冷速度-1000K/psの条件のもとで、界面ダングリングボンドのない急峻なアモルファスSiO/SiC界面構造を生成し、エネルギー準位や電荷分布等が導出できた。また、中規模モデルを用いて界面酸化反応の模擬計算を行い、酸素分子が連続して界面に達した場合に起こるSiO/SiC界面の酸化過程の第一原理分子動力学計算を世界で初めて成功させた。
/SiC界面構造の第一原理計算による生成,2宮下 敦巳; 大沼 敏治*; 岩沢 美佐子*; 土田 秀一*; 吉川 正人
no journal, ,
Siに比べ優れた物理特性を持つSiCを用いた半導体デバイスは、従来の半導体デバイスでは動作が困難な極限環境下で用いられる素子として期待されている。しかしSiCと酸化膜の界面には、SiCデバイスのチャネル移動度低下の原因となる界面欠陥が多く存在しており、その欠陥構造と素子特性との関連性を追求することは非常に重要である。本研究では実際のデバイスでの酸化膜界面を模擬するため、アモルファスSiO/SiC界面を計算機上に構築し、内在する欠陥構造の電子状態を算出することで、それが界面電気特性に与える影響を追求している。アモルファスSiO/SiC界面構造の生成は、第一原理分子動力学計算コードを用いた加熱・急冷計算法にて行った。1017原子界面構造モデルに対して、4000K 3psの固定終端加熱,終端解放後,3500K2psの継続加熱,-2000K/psで室温までの急冷を行った。生成したSiO層の動径分布関数を評価したところ、Si-O結合距離は0.165nm,Si-O-Si結合角は135,O-Si-O結合角は109と得られ、かつ、局所密度分布からも良好なアモルファス特性を示していることが確かめられた。
3psの固定終端加熱,終端解放後,3500K2psの継続加熱,-2000K/psで室温までの急冷を行った。生成したSiO層の動径分布関数を評価したところ、Si-O結合距離は0.165nm,Si-O-Si結合角は135,O-Si-O結合角は109と得られ、かつ、局所密度分布からも良好なアモルファス特性を示していることが確かめられた。
/4H-SiC(0001)界面における酸素解離反応の活性化エネルギー大沼 敏治*; 宮下 敦巳; 岩沢 美佐子*; 吉川 正人; 土田 秀一*
no journal, ,
ワイドギャップ半導体であるSiCはSi同様熱酸化により絶縁膜を作製できるため、次世代のMOS型パワーデバイスとして有望である。しかしSiC/SiO界面においては、Si/SiO界面に比べて界面トラップ密度が高いことや、MOSデバイスのチャンネル移動度が低いことが知られている。このような性能劣化の原因となる欠陥構造の形成過程の解明のためには、SiC/SiO界面の熱酸化過程のメカニズムを明らかにすることが重要であることから、われわれは第一原理分子動力学シミュレーションを用いた界面酸化模擬計算を進めている。界面へ酸素分子を導入して計算を行ったところ、一番目の酸素分子は界面中のSi原子と結合解離し、反応の活性化エネルギーは1.8eVとSiの熱酸化における値とほぼ同じ大きさであった。二番目の酸素分子は界面Si原子と結合・解離し、反応の活性化エネルギーは3.6eVであった。界面Si原子との酸化反応の活性化エネルギーが大きいことから、直接界面Si原子を酸化するよりはSiO層中のSi原子と反応しやすいことがわかる。三番目の酸素分子の解離反応の活性化エネルギーはほぼ0eVであり、酸化が進みダングリングボンドが多数存在すると解離反応が容易に起きることがわかった。
とUOにおける原子間力ポテンシャルの特性の研究中村 仁一; 金田 保則*; Chen, Y.*; Geng, H.*; 岩沢 美佐子*; 大沼 敏治*; 木下 幹康
no journal, ,
照射環境下で核燃料内に発生するさまざまな欠陥構造の理論的解明には、分子動力学法を用いた解析が有効である。特に面状欠陥の発生に着目し、この計算による再現を目標とした、UO並びに模擬物質としてのCeOの原子間ポテンシャルの開発・特性解析の現状について述べる。
とCeO中の欠陥生成の第一原理エネルギー論的研究中村 仁一; Chen, Y.*; 岩沢 美佐子*; 大沼 敏治*; 金田 保則*; Geng, H.*; 木下 幹康
no journal, ,
核燃料であるUOと加速器実験の模擬材料としてのCeOの物性・構造変化を理解、比較するため、核分裂生成物であるXe原子を含む種々の欠陥系の第一原理計算を行う、二つの系において電子構造,点欠陥生成エネルギーと格子緩和効果を求めた。さらに、有限温度で各種点欠陥濃度を計算し、両系における欠陥生成特性と相違性を調べた。
における酸素格子間原子配置の第一原理計算,2; 量体と平面配置中村 仁一; Geng, H.*; Chen, Y.*; 金田 保則*; 岩沢 美佐子*; 大沼 敏治*; 木下 幹康
no journal, ,
The stability and energetics of Oxygen defect configrations in UO are studied by first principles method. The results show that, a little unexpected, the planar distribution favourable of oxygen interstitials and the possibility to form O-O dimer in UO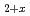
界面構造の第一原理計算による生成,3宮下 敦巳; 大沼 敏治*; 岩沢 美佐子*; 土田 秀一*; 吉川 正人
no journal, ,
第一原理分子動力学計算コードを用いた加熱・急冷計算によってアモルファス界面構造を構築し、電子状態が界面電気特性に与える影響を理論的側面から追求した。1017原子界面構造モデルに対して、冷却速度を従来用いていた-2000K/psから、-1000K/ps, -500K/psと変化させた所、室温冷却後の全エネルギーは-2000K/psの時を基準にして、それぞれ-7.2eV, -13.9eV低くなり安定化した。また、SiO層中でのSi-O-Si結合角も、135から、それぞれ137, 140と広くなり、よりシリカガラスでのSi-O-Siの結合角(145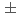 10)に近くなった。これらの結果から、冷却速度が遅い方が安定で実デバイスに近い界面構造を得られることがわかった。
10)に近くなった。これらの結果から、冷却速度が遅い方が安定で実デバイスに近い界面構造を得られることがわかった。
/4H-SiC(11-20)原子構造モデル宮下 敦巳; 大沼 敏治*; 土田 秀一*; 吉川 正人
no journal, ,
SiC半導体デバイスは、従来のデバイスでは動作が困難な極限環境でも使えるデバイスとして期待されている。SiC MOS-FETにおいては、ゲート酸化膜とSiC結晶との界面に多くの欠陥が存在しておりデバイス性能を劣化させている。実験的にそれらを解析することは困難であるため、原子構造モデルを計算機上に生成することで欠陥準位の同定を目指している。近年、4H-SiC(11-20)面で非常に良いチャネル移動度が得られることが報告されたことから、計算機上で当該面上にアモルファス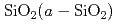 を生成し、その界面構造を評価した。まず、シリコン240個,炭素120個,酸素228個,水素48個を含む636原子による原子構造モデルに対して、4000K2ps及び3500K2psの加熱を行い層を溶融した後、-1000K/psの速度で室温までの急冷を行い、
を生成し、その界面構造を評価した。まず、シリコン240個,炭素120個,酸素228個,水素48個を含む636原子による原子構造モデルに対して、4000K2ps及び3500K2psの加熱を行い層を溶融した後、-1000K/psの速度で室温までの急冷を行い、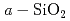 /4H-SiC(11-20)界面原子構造を生成した。中にはSiダングリングボンド、3配位のO等の欠陥が存在した。界面はほとんどがSi-O結合で接続していたが、他にもSi-Si結合,C-O結合,Si及びCダングリングボンドが観察された。
/4H-SiC(11-20)界面原子構造を生成した。中にはSiダングリングボンド、3配位のO等の欠陥が存在した。界面はほとんどがSi-O結合で接続していたが、他にもSi-Si結合,C-O結合,Si及びCダングリングボンドが観察された。
界面の生成; 温度条件による界面構造の違い宮下 敦巳; 大沼 敏治*; 岩沢 美佐子*; 土田 秀一*; 吉川 正人
no journal, ,
本研究では実際の界面に近い状態の原子構造を計算機上で生成し、その電子状態が界面電気特性に与える影響を理論的側面から追求している。実際のデバイス絶縁膜を模擬するために、加熱・急冷計算によりアモルファス界面構造を計算機上に構築し電子構造を決定した。計算はVASPコードを用いて行った。1017原子界面構造モデルに対して、4000Kで加熱後、-2000K/psで室温までの急冷を行い、生成した層の動径分布関数を評価した所、Si-O結合距離は0.165nm、O-Si-O結合角は109、Si-O-Si結合角は135と得られた。冷却速度を-1000K/ps, -500K/psと変化させた所、室温冷却後の全エネルギーは-2000K/psの時を基準にして、それぞれ-7.2eV, -13.9eV低くなった。また、Si-O-Si結合角も135から、それぞれ137, 140と広くなっており、よりシリカガラスでのSi-O-Siの結合角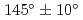 に近くなった。これらの結果から、冷却速度が遅い方がより安定で実デバイスに近い界面構造を得られることがわかった。
に近くなった。これらの結果から、冷却速度が遅い方がより安定で実デバイスに近い界面構造を得られることがわかった。
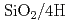 -
-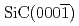 C面における熱酸化過程の第一原理分子動力学シミュレーション; Si面との違い
C面における熱酸化過程の第一原理分子動力学シミュレーション; Si面との違い大沼 敏治*; 宮下 敦巳; 岩沢 美佐子*; 吉川 正人; 土田 秀一*
no journal, ,
ワイドギャップ半導体であるSiCはSi同様熱酸化により絶縁膜を作製できるため次世代のMOS型パワーデバイスとして有望である。Si面と比較して酸化速度が10倍速い、チャネル移動度が大きいなど興味深い特徴を持つC面について、平面波基底のPAW法による第一原理分子動力学計算を行い、熱酸化過程の動的シミュレーションを行った。計算は地球シミュレーターで行った。界面モデルはスラブモデルを用い、界面の初期構造は加熱及び急冷法により作成した。界面モデルは急峻かつダングリングボンドのない綺麗な界面に酸化過程のきっかけとしてシリコン空孔を導入したものを用いた。酸化過程のシミュレーションは酸素分子を一つずつSiO層に追加することにより行った。酸素分子はSiO層中及びSiC界面のSi原子と反応し解離し、SiC界面のSi原子が酸化されることによりSi原子とC原子との結合が切れてCダングリングボンドが生成された。界面の炭素原子は酸化されてCO及びCO分子,C-C-O複合体が生成した。C面においてはSi面よりも生成したCO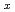 分子がCダングリングボンドと反応しにくく、拡散しやすいことがわかった。
分子がCダングリングボンドと反応しにくく、拡散しやすいことがわかった。
and CeOChen, Y.*; 金田 保則*; Geng, H. Y.*; 木下 幹康; 岩沢 美佐子*; 大沼 敏治*; 中村 仁一
no journal, ,
重照射下でのUOの欠陥構造やその回復過程を理解するために、UOと加速器実験で模擬燃料材とするCeOについて、核分裂生成物であるXeを含む種々の点欠陥に関する第一原理計算を行った。その結果、いずれの系においても、酸素点欠陥のふるまい・特徴は異なるものの添加されたXe原子の挙動や熱力学的特性は同じ傾向を示した。また、非化学量論状態にある2つの系における酸素クラスターの安定性について計算を行い、実効的な酸素クラスター相互作用エネルギーを求め、イオン結晶と金属結晶の間の相互作用の違いを明らかにした。
