


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
徳満 駿*; 三島 卓己*; 松宮 正彦*; 佐々木 祐二
Journal of Molecular Liquids, 414, Part A, p.126150_1 - 126150_8, 2024/11
希土類元素の電析による回収工程に用いる溶媒としてイオン液体が期待されている。リン酸系イオン液体は粘性が低く、高温で安定であるため、有望な溶媒である。イオン液体中の電析反応は金属イオンの溶存状態の影響を受けるが、回収工程で想定される高温条件での溶存状態と熱力学パラメータについては知見が不足している。そこで、本研究ではリン酸系イオン液体bis(trifluoromethyl sulfonyl)amide(TFSA)を用いて、ラマン分光により希土類元素(Pr, Nd, Tb, Dy)の溶存状態を調査し、温度依存性を取得することで熱力学パラメータを決定した。その結果、いずれの希土類元素もTFSAの5配位錯体を形成しやすく、錯体構造中ではTFSAはcis-異性体がより安定となることが分かった。加えて、密度汎関数法に基づく量子化学計算により錯体の結合エネルギー等を求め、実験により求めた熱力学的特性と矛盾しないことを確認した。
伊藤 孝; 髭本 亘; 下村 浩一郎*
Physical Review B, 108(22), p.224301_1 - 224301_11, 2023/12
被引用回数:6 パーセンタイル:59.15(Materials Science, Multidisciplinary)In positive muon spin rotation and relaxation (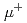 SR) spectroscopy, positive muons () implanted into solid oxides are conventionally treated as immobile spin-probes at interstitial sites below room temperature. This is because each is thought to be tightly bound to an oxygen atom in the host lattice to form a muonic analogue of the hydroxy group. On the basis of this concept, anomalies in SR spectra observed in oxides have been attributed in most cases to the intrinsic properties of host materials. On the other hand, global diffusion with an activation energy of
SR) spectroscopy, positive muons () implanted into solid oxides are conventionally treated as immobile spin-probes at interstitial sites below room temperature. This is because each is thought to be tightly bound to an oxygen atom in the host lattice to form a muonic analogue of the hydroxy group. On the basis of this concept, anomalies in SR spectra observed in oxides have been attributed in most cases to the intrinsic properties of host materials. On the other hand, global diffusion with an activation energy of  0.1~eV has been reported in some chemically-substituted perovskite oxides at cryogenic temperatures, although the reason for the small activation energy despite the formation of the strong O
0.1~eV has been reported in some chemically-substituted perovskite oxides at cryogenic temperatures, although the reason for the small activation energy despite the formation of the strong O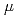 bond has not yet been quantitatively understood. In this study, we investigated interstitial diffusion in the perovskite oxide lattice using KTaO
bond has not yet been quantitatively understood. In this study, we investigated interstitial diffusion in the perovskite oxide lattice using KTaO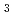 cubic perovskite as a model system. We used the SR method and density functional theory calculations along with the harmonic transition state theory to study this phenomenon both experimentally and theoretically. Experimental activation energies for global diffusion obtained below room temperature were less than a quarter of the calculated classical potential barrier height for a bottleneck transfer path. The reduction in the effective barrier height could be explained by the harmonic transition state theory with a zero-point energy correction; a significant difference in zero-point energies for at the positions in the O bonding equilibrium state and a bond-breaking transition state was the primary cause of the reduction. This suggests that the assumption of immobile in solid oxides is not always satisfied since such a significant decrease in diffusion barrier height can also occur in other oxides.
cubic perovskite as a model system. We used the SR method and density functional theory calculations along with the harmonic transition state theory to study this phenomenon both experimentally and theoretically. Experimental activation energies for global diffusion obtained below room temperature were less than a quarter of the calculated classical potential barrier height for a bottleneck transfer path. The reduction in the effective barrier height could be explained by the harmonic transition state theory with a zero-point energy correction; a significant difference in zero-point energies for at the positions in the O bonding equilibrium state and a bond-breaking transition state was the primary cause of the reduction. This suggests that the assumption of immobile in solid oxides is not always satisfied since such a significant decrease in diffusion barrier height can also occur in other oxides.
 inferred from muon study
inferred from muon study門野 良典*; 平石 雅俊*; 岡部 博孝*; 幸田 章宏*; 伊藤 孝
Journal of Physics; Condensed Matter, 35(28), p.285503_1 - 285503_13, 2023/07
被引用回数:1 パーセンタイル:8.99(Physics, Condensed Matter)Magnesium hydride has great potential as a solid hydrogen (H) storage material because of its high H storage capacity of 7.6 wt%. However, its slow hydrogenation and dehydrogenation kinetics and the high temperature of 300 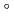 C required for decomposition are major obstacles to small-scale applications such as automobiles. The local electronic structure of interstitial H in MgH is an important fundamental knowledge in solving this problem, which has been studied mainly based on density functional theory (DFT). However, few experimental studies have been performed to assess the results of DFT calculations. We have therefore introduced muon (Mu) as pseudo-H into MgH and investigated the corresponding interstitial H states by analyzing their electronic and dynamical properties in detail. As a result, we observed multiple Mu states similar to those observed in wide-gap oxides, and found that their electronic states can be attributed to relaxed-excited states associated with donor/acceptor levels predicted by the recently proposed ambipolarity model. This provides an indirect support for the DFT calculations on which the model is based via the donor/acceptor levels. An important implication of the muon results for improved hydrogen kinetics is that dehydrogenation, serving as a reduction for hydrides, stabilises the interstitial H
C required for decomposition are major obstacles to small-scale applications such as automobiles. The local electronic structure of interstitial H in MgH is an important fundamental knowledge in solving this problem, which has been studied mainly based on density functional theory (DFT). However, few experimental studies have been performed to assess the results of DFT calculations. We have therefore introduced muon (Mu) as pseudo-H into MgH and investigated the corresponding interstitial H states by analyzing their electronic and dynamical properties in detail. As a result, we observed multiple Mu states similar to those observed in wide-gap oxides, and found that their electronic states can be attributed to relaxed-excited states associated with donor/acceptor levels predicted by the recently proposed ambipolarity model. This provides an indirect support for the DFT calculations on which the model is based via the donor/acceptor levels. An important implication of the muon results for improved hydrogen kinetics is that dehydrogenation, serving as a reduction for hydrides, stabilises the interstitial H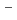 state.
state.
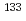 Cs NMR chemical shift of clay minerals via machine learning and DFT-GIPAW calculations
Cs NMR chemical shift of clay minerals via machine learning and DFT-GIPAW calculations大窪 貴洋*; 武井 滉洋*; 舘 幸男; 深津 勇太; 出口 健三*; 大木 忍*; 清水 禎*
Journal of Physical Chemistry A, 127(4), p.973 - 986, 2023/02
被引用回数:7 パーセンタイル:65.10(Chemistry, Physical)粘土鉱物へのCsの吸着サイトの特定は、環境化学の分野で研究されてきた。核磁気共鳴(NMR)実験によって、吸着されたCsの局所構造を直接観察することが可能である。固体NMR実験から得られたCsのNMRパラメータは、吸着されたCsの局所的な構造に敏感である。しかしながら、NMRデータだけからCsの吸着位置を決定することは困難であった。本研究では、機械学習と実験的に観察されたケミカルシフトを組み合わせることにより、粘土鉱物に吸着されたCsの吸着位置を特定するためのアプローチについて提示する。原子配置の記述子とNMRによるケミカルシフトの第一原理計算結果を関連付けて評価する機械学習の線形リッジ回帰モデルを構築した。これにより、原子配置の構造データからCsのケミカルシフトを高速で計算することが可能となった。機械学習モデルによって、実験的に観察された化学シフトから逆解析を行うことにより、Cs吸着位置を導き出すことが可能になる。
佐々木 祐二; 中瀬 正彦*; 金子 政志; 小林 徹; 竹下 健二*; 松宮 正彦*
Analytical Sciences, 5 Pages, 2023/00
被引用回数:0 パーセンタイル:0.00(Chemistry, Analytical)Ru抽出に関して、XANESスペクトルとDFT計算による理論的な検討を行った。各種鉱酸からMIDOA(メチルイミノジオクチルアセトアミド)によるRu分配比はHCl 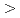 HSO
HSO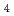 HNO HClOの順になった。XANESスペクトルの結果から、HClから抽出したRuの原子価は低く、有機相中でアニオン性のRuの存在とこれによるイオン対抽出を示唆した。抽出剤相互のRu分配比の比較結果(NTAamide MIDOA IDOA)はDFT計算のHOMO, LUMO間のエネルギー差と一致した。これは抽出能力と反応熱量の間に重要な関連があることを示した。
HNO HClOの順になった。XANESスペクトルの結果から、HClから抽出したRuの原子価は低く、有機相中でアニオン性のRuの存在とこれによるイオン対抽出を示唆した。抽出剤相互のRu分配比の比較結果(NTAamide MIDOA IDOA)はDFT計算のHOMO, LUMO間のエネルギー差と一致した。これは抽出能力と反応熱量の間に重要な関連があることを示した。
において観測される局在電子の起源について伊藤 孝
めそん, (56), p.21 - 26, 2022/09
近年、水素原子・イオン打ち込み技術の発展に伴い、熱力学的固溶限を超えて大量の水素を固体材料の格子間隙に強制的に導入できるようになってきた。これにより、絶縁体のホストを金属化するなどの振り幅の大きい物性制御が可能になりつつある。それとともに、低濃度ドープでは認知されなかった余剰電子の奇妙な振る舞いが明かになってきた。本稿では、代表的な電子材料の1つであるSrTiOを水素照射により金属化した際に、遍歴電子とともに局在電子が存在する痕跡が観測されたという事実に注目し、この局在電子の起源についてSR実験と密度汎関数理論(DFT)計算の結果に基づいて議論する。
Tang, J.*; Seo, O.*; Rivera Rocabado, D. S.*; 小板谷 貴典*; 山本 達*; 難波 優輔*; Song, C.*; Kim, J.*; 吉越 章隆; 古山 通久*; et al.
Applied Surface Science, 587, p.152797_1 - 152797_8, 2022/06
被引用回数:13 パーセンタイル:67.51(Chemistry, Physical)水素貯蔵材料として重要な立方体形状Pdナノ粒子の水素吸収と拡散メカニズムをX線光電子分光とDFT計算を用いて調べた。表面領域では粒子の大きさによらず、ほぼ同様の水素吸収挙動を示した。四面体サイトよりも八面体サイトの水素占有率が大きいことがわかった。表面の乱れによってPd-H結合が弱くなるため、小さいサイズのPdナノ粒子に吸収された水素原子は、より活発に粒子内部に拡散することが分かった。これが低水素圧での水素吸着に重要な役割を果たしている。
鈴木 知史; 逢坂 正彦; 中桐 俊男
Proceedings of Joint International Conference on Supercomputing in Nuclear Applications + Monte Carlo 2020 (SNA + MC 2020), p.131 - 136, 2020/10
人形峠におけるウラン移行挙動評価に資するため、ウラン鉱物の安定性評価をDFT計算により進めている。その第一段階として、主要なウラン鉱物で構造の詳細が明らかでない人形石の結晶構造と単位胞の構成を検討した。人形石の結晶構造の評価のため、XRD解析コードを用いて既報の人形石のXRDパターンを解析した。その結果、U原子間の距離は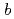 軸に沿って
軸に沿って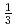 となった。そこで、3個のUまたはCa原子が軸に沿って並んだ[CaU(PO
となった。そこで、3個のUまたはCa原子が軸に沿って並んだ[CaU(PO )
) ]
] の構造を構成し、この構造を初期構造としてDFT計算により構造最適化を実施した。その結果、得られた構造の理論XRDパターンは、人形石のXRDパターンと同様に、30
の構造を構成し、この構造を初期構造としてDFT計算により構造最適化を実施した。その結果、得られた構造の理論XRDパターンは、人形石のXRDパターンと同様に、30  付近に最大のピークがあった。これにより、人形石の構造は[CaU(PO)]を基礎とする構造であると考えられる。
付近に最大のピークがあった。これにより、人形石の構造は[CaU(PO)]を基礎とする構造であると考えられる。
Miradji, F.; 鈴木 知史; 中島 邦久; 逢坂 正彦
Journal of Physics and Chemistry of Solids, 136, p.109168_1 - 109168_9, 2020/01
被引用回数:4 パーセンタイル:19.35(Chemistry, Multidisciplinary)Under the scope of Fukushima Daiichi Nuclear Power Station (1-F) severe accident (SA), Cs retention is of high interest as its impacts Cs distribution, decommissioning and dismantling work of the reactor. To derive consistent and appropriate models for such process, accurate thermodynamic properties of Cs chemisorbed species are required by the SA analysis codes. In particular, for CsFeSiO, a newly identified Cs chemisorbed species under conditions similar to 1-F SA, the thermodynamic data are unknown in literature. We propose in this work the obtention of the fundamental properties of this substance by theoretical approaches. The consistency and appropriateness of derived computational methodology have been investigated by calculating the thermodynamic properties of relatively known Cs-Si-O substances. It was found that our computational methodology provides excellent agreement with literature data lying between 1-4% for the formation energy, 1-5% for standard entropy and heat capacity. The thermodynamic properties of CsFeSiO in function of temperature have been estimated for the first time using harmonic and quasi-harmonic approximations, values being consistent with both methodologies.
足立 将晶*; 石附 茂*; 小笠原 忍*; 久米 悦雄; 箭竹 陽一*; 根本 俊行*; 川崎 信夫*; 川井 渉*
JAERI-Data/Code 2000-043, 220 Pages, 2001/02
 JAERI-Data-Code-2000-043.pdf:7.39MB
JAERI-Data-Code-2000-043.pdf:7.39MB
本報告書は、平成11年度に計算科学技術推進センター情報システム管理課で行った原子力コードの高速化作業のうち、VPP500(一部SX-4含む)におけるベクトル化/並列化作業部分について記述したものである。原子力コードの高速化作業は、平成11年度に18件行われた。これらの作業内容は、今後同種の作業を行ううえでの参考となりうるよう、作業を大別して「ベクトル/並列化編」,「スカラ並列化編」及び「移植編」の3分冊にまとめた。本報告書の「ベクトル/並列化編」では、JAMコード及び3次元熱流体解析コードSTREAMを対象に実施したベクトル化作業について、相対論的分子軌道法コードRSCAT,相対論的密度汎関数法コードRDFT及び高速3次元中性子拡散ノート法コードMOSRA-Lightを対象に実施したベクトル並列化作業について記述している。
Bastug, T.; 平田 勝; Varga, S.*; Fricke, B.*; Erkoc, S.*; 向山 毅*
Advances in Quantum Chemistry, Volume 37, p.353 - 364, 2001/00
相対論密度汎関数法(RDFT)を用いて、金(Au)2原子分子の構造最適化計算を行い、ポテンシャルエネルギー曲線を作成した。このポテンシャルを用いてAu3個から555個のクラスターの分子動力学(MD)シミュレーションを行った。その結果、Au13個で構成されるクラスターが最も対称性が良く(Ih対称)安定に存在する可能性の高いことがわかった。
平田 勝; Bastug, T.; 館盛 勝一
日本原子力学会誌, 42(10), p.1104 - 1108, 2000/10
被引用回数:1 パーセンタイル:11.79(Nuclear Science & Technology)相対論密度汎関数(RDFT)法を用いて硝酸プルトニル・2水和物のPu=O(プルトニウム酸素)結合及びPu-OH2結合におけるプルトニウム原子と配位子酸素原子間距離の構造最適化を行った。計算によって求められた原子間距離は実験的に報告されている原子間距離と良好な一致を示した。得られたポテンシャル曲線からは、Pu-OH2結合と比較してPu=O結合が非常に強いことを示した。また、安定構造における硝酸プルトニル・2水和物の電子状態を解析した結果、Pu=O結合の強さの原因は、プルトニウムの6d軌道と酸素の2p軌道との強い共有結合性に伴うものであることを明らかにした。また、これらの共有結合成分はPu-OH2結合ではそれほど顕著ではなかった。
Miradji, F.; 鈴木 恵理子; 西岡 俊一郎; 鈴木 知史; 中島 邦久; 大石 佑治*; 牟田 浩明*; 黒崎 健*; Do, Thi Mai Dung*; 逢坂 正彦
no journal, ,
Cesium (Cs) chemisorption model has been developed for estimation of Cs distribution in the reactor pressure vessels of the Fukushima Dai-ichi Nuclear Power Station. This model treats the chemical features of the Cs chemisorption process, which requires accurate thermodynamic data for the Cs chemisorbed compounds. For this purpose, we compared thermodynamic properties of major formed/predicted Cs silicates, Cs-(Fe)-Si-O, obtained from DFT calculations with ones from experimental data to mutually estimate the accuracy of both methodologies. As the results, a great accordance is found for the thermodynamic assessment of CsSiO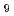 and CsFeSiO substances from all sources.
and CsFeSiO substances from all sources.
Miradji, F.; 鈴木 知史; 中島 邦久; 逢坂 正彦
no journal, ,
In the framework of FDNPS severe accident (SA), Cesium (Cs) retention phenomena are of high interest due to their impact on the decommissioning and dismantling work. Current SA analysis codes present large uncertainties in the estimation of Cs retention at reactor coolant system regions, due in part to the lack of knowledge in Cs chemisorption processes onto stainless steel (SS) surfaces. To improve and extend knowledge on Cs chemisorption mechanisms with SS, we investigated potential Cs-(Fe)-Si-O formed species by means of experimental and theoretical approaches. In this study, we will present the derivation of Cs-(Fe)-Si-O substances structural and thermodynamic properties from DFT calculations and the validation of developed computational methodology through comparisons with available literature data. In particular, the thermodynamic properties of newly characterized Cs chemisorbed species in our experimental tests are provided for the first time.
木下 了磨; 佐々木 祐二; 金子 政志; 松宮 正彦*; 新奥 孝太*; 城石 英伸*
no journal, ,
塩酸溶液中で多くの金属イオンはアニオン種で安定に存在する。NTAamideを代表とする3級アミノ窒素を含むジアミノ抽出剤は酸性溶液中でプロトネーションを起こし、カチオン性の抽出剤となる。本研究では、多種の金属塩化物アニオンとカチオン種のNTAamide(C6)のイオン対抽出反応について調査を行った。併せて、金属塩化物錯体の安定度定数やDFT計算を組み合わせることによって分配比を予測し、実験値との比較を行った。
宮崎 康典; 渡部 創; 佐野 雄一; 小藤 博英; 竹内 正行; 石神 龍哉*; 江夏 昌志*; 佐藤 隆博*
no journal, ,
使用済燃料の再処理工程で発生する高レベル放射性廃棄物には、長寿命核種や発熱性核種であるマイナーアクチノイドが含まれており、これらを取り除くことで地層処分における廃棄物減容化や有害度低減に繋がる。そこで、抽出クロマトグラフィを用いたMAの選択的分離回収を検討している。これまでに、高分子ポリマーで塗布した多孔質SiO粒子にCMPO等の抽出剤を含浸させた吸着材を作製し、カラム運転中に想定される放射線分解物の特定及び挙動調査を行ってきた。本研究では、 線を模擬したTODGA/SiO-P吸着材の放射線耐性評価と理論計算による結合解離メカニズムの解明を目的に、Heイオンの照射試験を実施した。Heイオン照射に対して、酸の有無による抽出剤の分解生成物に違いが見られた。硝酸を加えてスラリー状態で照射した場合、アミドC-N結合の解離のみが起きたが、乾燥状態ではアミドC-N結合に加えてエーテルC-O結合の解離も見られた。この違いはプロトン付加した分子構造に依存すると考え、量子化学計算を用いて結合解離エネルギー(BDE)を求めた。フリーのDGAのエーテル結合のBDEは、アミド結合よりも小さい。一方、プロトン付加した構造では、エーテル結合はアミド結合よりもBDEが大きくなる。つまり、結合解離が起きにくいと予想された。この結果は実験で得られた結合解離パターンと傾向が一致した。今後、照射線量と吸着材の劣化生成量における相関を調査し、抽出クロマトグラフィに用いる吸着材の再利用性を含めた研究を行う。
線を模擬したTODGA/SiO-P吸着材の放射線耐性評価と理論計算による結合解離メカニズムの解明を目的に、Heイオンの照射試験を実施した。Heイオン照射に対して、酸の有無による抽出剤の分解生成物に違いが見られた。硝酸を加えてスラリー状態で照射した場合、アミドC-N結合の解離のみが起きたが、乾燥状態ではアミドC-N結合に加えてエーテルC-O結合の解離も見られた。この違いはプロトン付加した分子構造に依存すると考え、量子化学計算を用いて結合解離エネルギー(BDE)を求めた。フリーのDGAのエーテル結合のBDEは、アミド結合よりも小さい。一方、プロトン付加した構造では、エーテル結合はアミド結合よりもBDEが大きくなる。つまり、結合解離が起きにくいと予想された。この結果は実験で得られた結合解離パターンと傾向が一致した。今後、照射線量と吸着材の劣化生成量における相関を調査し、抽出クロマトグラフィに用いる吸着材の再利用性を含めた研究を行う。
鈴木 智也*; 塩飽 秀啓; 尾形 剛志*; 谷田 肇; 小林 徹; 矢板 毅; 成田 弘一*
no journal, ,
塩酸水溶液中における三価白金属イオンは、抽出不活性であることが知られている。我々のグループでは、トリオクチルアミンとジヘキシルスルフィドの混合溶媒により、三価白金属イオンを抽出できることを見出している。今回はRu(III)を抽出するために、トリオクチルアミンとスルフィド系化合物の抽出性能をICP-AESにより定量し、錯体構造や錯形成を放射光XAFSを用いて検討した。DFT計算を行い、協同抽出メカニズムについても考察する。
Miradji, F.; 鈴木 知史; 中島 邦久; 逢坂 正彦
no journal, ,
Within the framework of Fukushima Daiichi NPS decommissioning work, the modelling of Cs chemisorption process onto reactor structure surfaces need to be improved, as models implemented in SA codes cannot reproduce the chemisorption amount under the 1F-SA chemical conditions. To build Cs chemisorption model based on the mass transfer theory, the thermal properties of identified Cs-Fe-Si-O chemisorbed species are needed. We present in this work the derivation of CsFeSiO and CsSiO properties by quantum chemical methodologies. In particular. Density Functional Theory (DFT) was applied to obtain the geometric, electronic and energetic properties of target species. Phonon calculations in the harmonic approximation were performed to derive the thermodynamic data.
矢板 毅
no journal, ,
Three years have passed since Fukushima Dai-ichi Nuclear Power Plant (FDNP) Accident, a serious contamination problem still remains in Fukushima. Especially, the 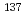 Cs in soil is one of the main sources to increase the air dose in Fukushima. Therefore, Japanese and local governments promotes the decontamination projects in which contaminated surface soil is removed and then, is moved it to interim storage area. Since soil waste is extremely large and space of an interim storage is limited, suitable waste reduction method would be required, also in anticipation of final disposal of the waste after about 30 years. On these backgrounds, we have performed elucidation of the sorption-desorption behaviors of Cs on clay minerals by XAFS, STXM, and theoretical calculation methods, aiming at soil waste volume reduction in Fukushima.
Cs in soil is one of the main sources to increase the air dose in Fukushima. Therefore, Japanese and local governments promotes the decontamination projects in which contaminated surface soil is removed and then, is moved it to interim storage area. Since soil waste is extremely large and space of an interim storage is limited, suitable waste reduction method would be required, also in anticipation of final disposal of the waste after about 30 years. On these backgrounds, we have performed elucidation of the sorption-desorption behaviors of Cs on clay minerals by XAFS, STXM, and theoretical calculation methods, aiming at soil waste volume reduction in Fukushima.
