


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
中川 洋; 米谷 佳晃*; 中島 健次; 河村 聖子; 菊地 龍弥*; 稲村 泰弘; 片岡 幹雄*; 河野 秀俊*
JPS Conference Proceedings (Internet), 33, p.011101_1 - 011101_6, 2021/03
5'CGCG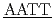 CGCG'3 and 5'CGCG
CGCG'3 and 5'CGCG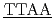 CGCG'3のDNAについて、軽水と重水のコントラストを利用した中性子準弾性散乱による水和水ダイナミクスを測定した。この2つのDNAは計算機によってそれぞれ硬い分子と柔らかい分子であることが分かっている。どちらの配列も約240KにDNAと水和水のどちらも動力学転移が観測された。転移温度以上では、水和水の平均自乗変位は硬い配列の方が小さかった。また水和水の緩和時間は硬いほうが長かった。ピコ秒時間スケールの水和水ダイナミクスは配列依存的なDNAの硬さと関係していることを示唆した。
CGCG'3のDNAについて、軽水と重水のコントラストを利用した中性子準弾性散乱による水和水ダイナミクスを測定した。この2つのDNAは計算機によってそれぞれ硬い分子と柔らかい分子であることが分かっている。どちらの配列も約240KにDNAと水和水のどちらも動力学転移が観測された。転移温度以上では、水和水の平均自乗変位は硬い配列の方が小さかった。また水和水の緩和時間は硬いほうが長かった。ピコ秒時間スケールの水和水ダイナミクスは配列依存的なDNAの硬さと関係していることを示唆した。
米谷 佳晃*; 中川 洋
Chemical Physics Letters, 749, p.137441_1 - 137441_5, 2020/06
被引用回数:4 パーセンタイル:13.28(Chemistry, Physical)DNAのMDシミュレーションによりDNAの主鎖の水素原子の溶媒露出状態を計算した。計算された溶媒露出状態は、これまでの実験的研究で報告されているOHラジカルとの反応性と良い相関がみられた。これは、放射線に対する異なるDNAの反応性が、主にそれぞれの水素原子の溶媒露出状態の違いが原因であることを示している。過去に計算された溶媒露出表面積の計算と比較すると、今回の計算はわずかに良い結果をもたらしている。これは、静電相互作用とDNAの構造揺らぎのようなより現実の分子状態を反映した解析が重要であることを示唆する。
米谷 佳晃
Journal of Chemical Physics, 143(4), p.044506_1 - 044506_9, 2015/07
被引用回数:16 パーセンタイル:52.80(Chemistry, Physical)Different ion pairs exhibit different dissociation kinetics: however, while the nature of this process is vital for understanding various molecular systems, the underlying mechanism remains unclear. In this study, to examine the origin of different kinetic rate constants for this process, molecular dynamics simulations were conducted for LiCl, NaCl, KCl, and CsCl in water. The results showed substantial differences in dissociation rate constant. Analysis of the free-energy landscape with a solvent reaction coordinate and subsequent rate component analysis showed that the differences in these rate constants arose predominantly from the variation in solvent-state distribution between the ion pairs. The formation of a water-bridging configuration, in which the water molecule binds to an anion and a cation simultaneously, was identified as a key step in this process: water-bridge formation lowers the related dissociation free-energy barrier, thereby increasing the probability of ion-pair dissociation. Consequently, a higher probability of water-bridge formation leads to a higher ion-pair dissociation rate.
中川 洋; 米谷 佳晃; 中島 健次; 河村 聖子; 菊地 龍弥; 稲村 泰弘; 片岡 幹雄; 河野 秀俊
Physical Review E, 90(2), p.022723_1 - 022723_11, 2014/08
被引用回数:10 パーセンタイル:52.58(Physics, Fluids & Plasmas)CGCGAATTCGCGとCGCGTTAACGCGの柔軟性の異なる二つの配列のDNAの分子シミュレーションと中性子準弾性散乱実験を行った。前者は硬く、後者は柔らかいことが知られている。両方の配列のDNAで、200-240Kに動力学転移が見られた。DNA配列依存的なダイナミクスを調べるために、転移温度以上でDNAと水和水のダイナミクスを分子シミュレーションと中性子準弾性散乱によって調べた。12merDNAの真ん中の4merについて、AATTはTTAAと比べて揺らぎの振幅が小さく、緩和時間が長いことが分かった。これはATステップの方が、TAステップよりも、速度論的に安定であることを示唆している。配列依存的な局所的な塩基対のダイナミクスは、DNAと水和水の間の水素結合ダイナミクスと相関がある。配列依存的なDNAの塩基対の揺らぎは動力学転移温度以上で現れる。これらの結果を総合すると、DNAの柔軟性は塩基対の局所的なダイナミクスと関係があり、DNAのマイナー溝に存在する水和水とカップルしていると結論付けた。
米谷 佳晃; 河野 秀俊
Journal of Physical Chemistry B, 117(25), p.7535 - 7545, 2013/06
被引用回数:16 パーセンタイル:37.45(Chemistry, Physical)DNA-binding proteins recognize DNA sequences with at least two different binding modes, specific and nonspecific. Experimental structures of such complexes provide us a static view of the bindings. However, it is difficult to reveal further mechanisms of their target-site search and recognition only from static information because the transition process between the bound and unbound states is not clarified by static information. What is the difference between specific and nonspecific bindings? Here we performed adaptive biasing force molecular dynamics simulations with the specific and nonspecific structures of DNA-Lac repressor complexes to investigate the dissociation process. The resultant free-energy profiles showed that the specific complex has a sharp, deep well consistent with tight binding, whereas the non-specific complex has a broad, shallow well consistent with loose binding. The difference in the well depth, 5 kcal/mol, was in fair agreement with the experimentally obtained value and was found to mainly come from the protein conformational difference, particularly in the C-terminal tail. Comparison of the free-energy barrier for sliding,  8.7 kcal/mol, and that for dissociation (at least 16 kcal/mol) calculated in this study suggests that sliding is much preferred to dissociation.
8.7 kcal/mol, and that for dissociation (at least 16 kcal/mol) calculated in this study suggests that sliding is much preferred to dissociation.
米谷 佳晃; 河野 秀俊
アンサンブル, 14(4), p.182 - 186, 2012/10
生体分子表面は複雑であり、それゆえ水のダイナミクスは多様である。水分子の生体分子表面滞在時間は、数psから数100psまでさまざまであるが、その違いはどのようにして生じるのであろうか。筆者らは、さまざまな塩基配列を持つDNAを対象にした分子動力学シミュレーションから、DNAと水の水素結合様式とDNA表面の構造揺らぎが、水分子の滞在時間に関係していることを明らかにした。そこでは、Laage-Hynesにより示された水素結合組換えのメカニズムとの接点も明らかになった。今後、タンパク質の場合なども含め、水分子の滞在時間を統一的に理解し、表現していくためには、これまで示唆されてきた表面の形状と電気的性質の影響も考慮しなければならない。
米谷 佳晃; 河野 秀俊
Biophysical Chemistry, 160(1), p.54 - 61, 2012/01
被引用回数:21 パーセンタイル:53.72(Biochemistry & Molecular Biology)The lifetime during which a water molecule resides at the surface of a biomolecule varies according to the hydration site. What determines this variety of lifetimes? Despite many previous studies, there is still no uniform picture quantitatively explaining this phenomenon. Here we calculate the lifetime for a particular hydration pattern in the DNA minor groove, the water bridge, for various DNA sequences to show that the water-bridge lifetime varies from 1 to 300 ps in a sequence-dependent manner. We find that it follows 1/k (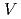
 )
)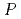
 , where and are two crucial factors, namely the probability of forming a specific hydrogen bond in which more than one donor atom participates, and the structural fluctuation of DNA, respectively. This relationship provides a picture of the water kinetics with atomistic detail and shows that water dissociation occurs when a particular hydrogen-bonding pattern appears.
, where and are two crucial factors, namely the probability of forming a specific hydrogen bond in which more than one donor atom participates, and the structural fluctuation of DNA, respectively. This relationship provides a picture of the water kinetics with atomistic detail and shows that water dissociation occurs when a particular hydrogen-bonding pattern appears.
樋口 真理子; 藤井 淳平*; 米谷 佳晃; 北尾 彰朗*; 郷 信広*
Journal of Structural Biology, 173(1), p.20 - 28, 2011/01
被引用回数:2 パーセンタイル:5.88(Biochemistry & Molecular Biology)ヌクレオシドdGTPと損傷したヌクレオシド8oxodGTPは構造が似ているが、修復酵素MutTは結合力の大きな差によって両者を区別している。本研究では分子動力学シミュレーションの手法を用いて、基質の小さな差を増幅し大きな結合力の差として認識する分子的な仕組みを解明した。MutTはdGMPとの結合時は基質周辺のループが開いている状態が安定だが、8oxodGMPとの結合時は基質周辺のループが閉じている状態が安定であることがわかった。このような結合構造の違いが結合力の大きな差を生み出していると考えられる。さらに、基質のH7とMutTの119番目の残基Asnをつなぐ水素結合が結合構造の違いに重要な役割を果たしていることがわかった。
米谷 佳晃; 河野 秀俊
揺らぎと生体機能, p.66 - 71, 2010/10
DNAの水和は、生命現象の根幹となる分子間相互作用の性質を理解するうえで重要である。近年、分子動力学シミュレーションにより、DNAのマイナーグルーブに現れる水和パターンに塩基配列依存性があること、また、水和水の挙動がDNAの構造揺らぎと密接に関係していることがわかってきた。本稿では、それらの知見について紹介するとともに、塩基配列によって異なる水和の性質がDNAとタンパク質,低分子の相互作用にどのように関連しているか議論する。
及川 雅隆; 米谷 佳晃
Biophysical Journal, 98(12), p.2974 - 2983, 2010/06
被引用回数:9 パーセンタイル:22.52(Biophysics)Are protein nonpolar cavities filled with water molecules ? Though many experimental and theoretical investigations have been done, particularly for the nonpolar cavity of IL-1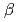 , the results are still conflicting. To study this problem from the thermodynamic point of view, we calculated hydration free energies of four protein nonpolar cavities by means of the molecular dynamics thermodynamic integration method. Besides the IL-1 cavity, we selected the three largest nonpolar cavities from the structural database, in view of the simulation result of Vaitheeswaran et al. [Proc. Natl. Acad. Sci. (2004)] showing that larger nonpolar cavities are more expected to be hydrated. The calculated free energy changes were all positive; hydration of the nonpolar cavities was energetically unfavorable for all four cases. Because hydration of smaller cavities should happen more rarely, we conclude that existing protein nonpolar cavities are not likely to be hydrated. Although a possibility remains for much larger nonpolar cavities, such cases are not found experimentally. We present a hypothesis to explain this: hydrated nonpolar cavities are quite unstable and the conformation could not be maintained.
, the results are still conflicting. To study this problem from the thermodynamic point of view, we calculated hydration free energies of four protein nonpolar cavities by means of the molecular dynamics thermodynamic integration method. Besides the IL-1 cavity, we selected the three largest nonpolar cavities from the structural database, in view of the simulation result of Vaitheeswaran et al. [Proc. Natl. Acad. Sci. (2004)] showing that larger nonpolar cavities are more expected to be hydrated. The calculated free energy changes were all positive; hydration of the nonpolar cavities was energetically unfavorable for all four cases. Because hydration of smaller cavities should happen more rarely, we conclude that existing protein nonpolar cavities are not likely to be hydrated. Although a possibility remains for much larger nonpolar cavities, such cases are not found experimentally. We present a hypothesis to explain this: hydrated nonpolar cavities are quite unstable and the conformation could not be maintained.
米谷 佳晃; 河野 秀俊
Biophysical Journal, 97(4), p.1138 - 1147, 2009/08
被引用回数:32 パーセンタイル:64.42(Biophysics)DNA deformability and hydration are both sequence dependent and are essential in specific DNA sequence recognition by proteins. However, their relationship is not well understood. Here, systematic molecular dynamics simulations of the 136 DNA sequences that differ from each other in the central tetramer revealed that sequence dependence of hydration is clearly correlated with that of deformability. We show that the feature of this correlation can be explained by the following four typical cases. Most of rigid base-pair steps are highly likely to form an ordered hydration pattern composed of one water molecule bridging between the bases of distinct strands, but there are a few exceptions which favor another ordered hydration composed of two water molecules participating in the bridge construction. Steps with medium deformability can make both of the hydration patterns with frequent transition. Highly flexible steps do not have any stable hydration pattern. The detailed picture of this correlation demonstrated that motions of hydration water molecules and of DNA bases were tightly coupled with each other at the atomic level. These results are helpful to understand the entropic contribution from water molecules in protein or drug binding and could be applied for predicting the binding sites.
米谷 佳晃*; 丸山 豊*; 平田 文男*; 河野 秀俊
Journal of Chemical Physics, 128(18), p.185102_1 - 185102_9, 2008/05
被引用回数:20 パーセンタイル:56.65(Chemistry, Physical)Although high-resolution, crystal structure analyses have given us a view of tightly bound water molecules on their surface, the structural data is still insufficient to capture the detailed configurations of water molecules around the surface of these biomolecules. Thanks to the invention of various computational algorithms, computer simulations can now provide an atomic view of hydration. Here we describe the apparent patterns of DNA hydration calculated using two different computational methods, MD simulation and 3D-RISM theory. This rigorous comparison showed that MD and 3D-RISM provide essentially similar hydration patterns when there is sufficient sampling time for MD and a sufficient number of conformations to describe molecular flexibility for 3D-RISM. This suggests that these two computational methods can be used to complement one another when evaluating the reliability of calculated hydration patterns.
石田 恒; 樋口 真理子; 米谷 佳晃*; 叶野 琢磨; 城地 保昌*; 北尾 彰朗*; 郷 信広
Annual Report of the Earth Simulator Center April 2005 - March 2006, p.237 - 240, 2007/01
地球シミュレータは従来にはない大規模生体超分子系の分子動力学シミュレーションを可能とする計算能力を持つ。そこで、われわれは数百万原子の生体超分子系を扱う大規模な分子動力学シミュレーションシステムSCUBA(旧名PABIOS)を開発している。SCUBAはPPM計算法,系のエネルギー,温度,圧力を一定に保つさまざまな時間積分アルゴリズム,系の原子分布異方性により引き起こされる並列化効率の悪化を防ぐための動的ロードバランスなど、最新のアルゴリズムを採用した計算性能に優れたシミュレーションシステムである。本年度は、約55万原子の系(RuvAB-Holliday分岐DNA)について分子動力学シミュレーションを実行した結果、地球シミュレータで360個のプロセッサを用いてもSCUBAはベクトル化率95%以上,並列化効率50%以上の優れた性能を達成することに成功した。そして、ホリデイジャンクションモデル(RuvA-Holliday分岐DNA)の長時間分子動力学シミュレーションを実行することで、Holliday分岐DNA結合タンパク質RuvAがDNAを組み換えるメカニズムを分子レベルで明らかにすることができた。
米谷 佳晃*; 河野 秀俊; 藤井 聡*; 皿井 明倫*; 郷 信広
Molecular Simulation, 33(1-2), p.103 - 107, 2007/01
被引用回数:5 パーセンタイル:16.09(Chemistry, Physical)5'AATT3'と5'TTAA3'の2種類の塩基配列のDNAについて分子動力学シミュレーションを行い、構造変化と水和の関係を調べた。シミュレーションから、5'AATT3'では、DNAは構造変化しにくく、水和水は構造化しやすいが、5'TTAA3'では、DNAは構造変化しやすく、水和水は構造化しやすいことが明らかになった。この結果に基づいてDNAの構造変化と水和の関係について議論した。
米谷 佳晃*
Journal of Chemical Physics, 124(20), p.204501_1 - 204501_11, 2006/05
被引用回数:60 パーセンタイル:87.98(Chemistry, Physical)分子動力学計算において分子間の相互作用をどこまで取り込むかは常に問題である。特に、距離の逆数で効くクーロン相互作用は遠距離まで作用する。しかし、計算効率を上げるため、第二水和水の距離、つまり、9A程度の距離で相互作用を打ち切った計算が多く行われてきた。本研究では、カットオフ距離を単純に長くすればシミュレーションが必ずしもより正確になるとはかぎらないことを見いだした。特に、水分子の配向は、カットオフ距離を長くすると逆に助長されることがあることを示した。さらに、このようなカットオフによるアーティファクトはスイッチング関数など滑らかな関数系を使うことで回避できることを示した。
石田 恒; 樋口 真理子; 米谷 佳晃*; 叶野 琢磨; 城地 保昌*; 北尾 彰朗*; 郷 信広
Annual Report of the Earth Simulator Center April 2004 - March 2005, p.241 - 246, 2005/12
地球シミュレータは従来にはない大規模超分子系の分子動力学シミュレーションを可能とする計算能力を持つ。そこで、われわれは数百万原子のシステムを扱う大規模な分子シミュレーション(PABIOS)を開発している。PABIOSは空間分割法を用いており高い並列化効率を持つ。PABIOSは系のエネルギー,温度,圧力を一定に保つさまざまな時間積分アルゴリズム,原子結合長を固定することにより時間ステップの増大を可能とするSHAKE, RATTLEアルゴリズムなどの高精度アルゴリズムを採用した計算性能に優れたシミュレーションシステムである。今回、系の原子分布の異方性による並列化効率の悪化がおこらないように、動的ロードバランスを開発した。ホリデイ分岐DNAとDNA結合蛋白質RuvA4量体の複合体が水中に存在する系(全系16万6千原子)について分子動力学シミュレーションを実行した結果、地球シミュレータで120個のプロセッサを用いてもPABIOSはベクトル化率95%以上,並列化効率50%以上の優れた性能を達成することに成功した。そして計算速度も昨年度と比べて約2倍程度向上した。
郷 信広; 米谷 佳晃*
生体の科学, 56(6), p.626 - 631, 2005/12
「蛋白質がわかる」ための理論と計算の役割について解説した。蛋白質の立体構造は、アミノ酸配列によって一意的に決まる。この背景にある仕組みを解明するという問題に関して、著者の一人は、かつて蛋白質の天然状態は整合性原理という原理を満たしていることが本質であることを指摘した。蛋白質のアミノ酸配列が生物進化の所産であることが、この原理成立の背景にある。理論は、このように問題の基本的構造を明らかにする役割を担っている。そして、近年、計算によって蛋白質を理解することが可能な時代に入りつつある。計算により蛋白質を理解するためには、蛋白質の構造区間のサンプリングと蛋白質を記述するポテンシャルエネルギー関数の2つの面の精度の向上が必要である。これらの2点について詳しく述べられている。
米谷 佳晃
Chemical Physics Letters, 406(1-3), p.49 - 53, 2005/04
被引用回数:52 パーセンタイル:83.97(Chemistry, Physical)18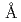 の長いカットオフ距離を用いて水の分子動力学シミュレーションを行ったところ、水の挙動に著しいアーティファクトが現れた。この結果から、カットオフ距離の増加によりシミュレーション結果が改善されるとは限らないことが判明した。さらに、これまでに長いカットオフ距離を用いて行われてきた水を含む生体分子系のシミュレーションにも予想外に大きいアーティファクトが存在していた可能性が示唆されている。
の長いカットオフ距離を用いて水の分子動力学シミュレーションを行ったところ、水の挙動に著しいアーティファクトが現れた。この結果から、カットオフ距離の増加によりシミュレーション結果が改善されるとは限らないことが判明した。さらに、これまでに長いカットオフ距離を用いて行われてきた水を含む生体分子系のシミュレーションにも予想外に大きいアーティファクトが存在していた可能性が示唆されている。
米谷 佳晃; 河野 秀俊
no journal, ,
「DNAと水」塩基配列に依存するDNAの性質,構造ダイナミクスと水和は、いずれも生体機能に関係する重要な性質であるが、これまで限られた配列についてしか議論されておらず、その配列依存性については統一的な解釈が導かれていなかった。そのため、われわれは21年度までに、すべての4塩基配列(136通り)について分子動力学計算による解析を行い、DNAのダイナミクスとマイナーグルーブの水和パターンの間に強い相関があることを見いだした。22年度はさらに水素結合持続時間を計算し、その配列依存性と水素結合解離のメカニズムを明らかにすることができたので、報告する。われわれの解析結果によると、水素結合持続時間は、1psから300psまで配列によりさまざまであり、t=1/kPmの関係に従う。ここで、Pmは多重水素結合状態の出現率であり、t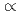 1/Pmは、水素結合の解離が、Pmの状態を経由して起こることを示唆する。配列間でのPmの違いが、水素結合持続時間tの違いに大きく関与していると考えられる。「DNAと蛋白質」DNA結合蛋白質がDNAに結合する際、はじめから認識配列に結合することはまれで、他の配列へ結合するなど幾つかの中間状態を経て認識配列に到達すると考えられている。中間状態の蛋白質は柔軟性の高い構造をとり、それが効率的配列探索に寄与していると考えられているが、未解明の点が多い。蛋白質の構造揺らぎが配列認識とどのように関係しているのかを明らかにするために、塩基配列を認識する過程における蛋白質の構造揺らぎと系の自由エネルギー変化の解析を進めている。その進捗状況について報告する。
1/Pmは、水素結合の解離が、Pmの状態を経由して起こることを示唆する。配列間でのPmの違いが、水素結合持続時間tの違いに大きく関与していると考えられる。「DNAと蛋白質」DNA結合蛋白質がDNAに結合する際、はじめから認識配列に結合することはまれで、他の配列へ結合するなど幾つかの中間状態を経て認識配列に到達すると考えられている。中間状態の蛋白質は柔軟性の高い構造をとり、それが効率的配列探索に寄与していると考えられているが、未解明の点が多い。蛋白質の構造揺らぎが配列認識とどのように関係しているのかを明らかにするために、塩基配列を認識する過程における蛋白質の構造揺らぎと系の自由エネルギー変化の解析を進めている。その進捗状況について報告する。
米谷 佳晃; 河野 秀俊
no journal, ,
DNAの構造揺らぎと水和は、いずれも生体機能に関係する重要な性質であるが、これまで限られた塩基配列についてしか議論されておらず、塩基配列による多様性についてのまとまった見解は得られていなかった。最近、136種類のDNA配列に対する分子動力学計算から、DNAの構造揺らぎの配列依存性が明らかになった。本研究では、同様の配列に対して水和パターンを調べ、両者を比較することで、DNAの構造揺らぎとマイナーグルーブの水和パターンの間に強い相関があることを見いだした。さらに、その関係に注目して、配列による両者の性質の多様性を、4つのタイプで簡潔に記述することに成功した。本研究を通じて明らかになった水和パターンの配列依存性が、蛋白質との相互作用の場面でどのように関係するのかという問題についても議論する。
