


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
 O
O -pentadentate planar ligands designed for the strongest and selective capture of uranium from seawater
-pentadentate planar ligands designed for the strongest and selective capture of uranium from seawater水町 匠*; 佐藤 みなみ*; 金子 政志; 竹山 知志*; 津島 悟*; 鷹尾 康一朗*
Inorganic Chemistry, 61(16), p.6175 - 6181, 2022/04
被引用回数:4 パーセンタイル:58.46(Chemistry, Inorganic & Nuclear)海水中のウランに対して強力かつ選択的に錯体を生成する平面五座配位子Hsaldian及びその誘導体を開発した。模擬海水条件において、saldian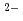 はウランに対して非常に高い安定度定数を示し、従来開発されてきたアミドキシム型配位子よりも10桁以上の錯生成能を達成した。海水中に存在する他の夾雑イオンに対するウランの選択性についても、良好な結果が得られた。
はウランに対して非常に高い安定度定数を示し、従来開発されてきたアミドキシム型配位子よりも10桁以上の錯生成能を達成した。海水中に存在する他の夾雑イオンに対するウランの選択性についても、良好な結果が得られた。
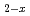 based on experimental data and density functional theory calculation result
based on experimental data and density functional theory calculation result渡部 雅; 中村 博樹; 鈴木 紀一; 町田 昌彦; 加藤 正人
Journal of the American Ceramic Society, 105(3), p.2248 - 2257, 2022/03
被引用回数:1 パーセンタイル:6.33(Materials Science, Ceramics)CeOのバンドギャップ,フレンケル欠陥生成エネルギー及び欠陥移動エネルギーを決定するため、DFTシュミュレーションによる評価を行った。バンドギャップ及びフレンケル欠陥生成エネルギーは欠陥平衡を解析するために使用した。欠陥平衡の酸素分圧依存性は酸素ポテンシャルの実験データとDFT計算に基づいて評価し、Brouwer図を導出した。フレンケル欠陥、電子-正孔対等の欠陥形成エネルギーを決定し、酸素拡散係数,電気伝導率,比熱容量及び熱伝導率の評価に用いた。これらの物性値のメカニズムについての理解を深めるため欠陥化学に基づく議論を行い、物性値の関係を系統的に記述した。
金子 政志
日本原子力学会誌ATOMO , 64(1), p.30 - 34, 2022/01
, 64(1), p.30 - 34, 2022/01
マイナーアクチノイドと希土類元素の分離は、高レベル放射性廃液の群分離プロセス開発において肝となる技術である。本稿では密度汎関数法という計算科学手法を用いて、溶媒抽出法によるマイナーアクチノイドと希土類元素の分離性能の再現や、その分離メカニズム解明に向けてアプローチした結果について解説する。
金子 政志; 佐々木 祐二; 和田 恵梨子*; 中瀬 正彦*; 竹下 健二*
Chemistry Letters, 50(10), p.1765 - 1769, 2021/10
被引用回数:0 パーセンタイル:0(Chemistry, Multidisciplinary)密度汎関数計算を用いて、マイナーアクチノイドとランタノイドの新規分離試薬の分子設計に向けて水溶液中におけるEu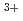 及びAm錯体の安定度定数を予想した。実験値の安定度定数の対数と計算した錯生成エンタルピーは、一次の決定係数R
及びAm錯体の安定度定数を予想した。実験値の安定度定数の対数と計算した錯生成エンタルピーは、一次の決定係数R
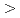 0.98で相関した。さらに、新規キレート配位子の安定度定数の予測を試み、ジエチレントリアミン五酢酸キレート配位子の誘導体が、酸性条件での使用やAm分離性能の観点から有用であることが示唆された。
0.98で相関した。さらに、新規キレート配位子の安定度定数の予測を試み、ジエチレントリアミン五酢酸キレート配位子の誘導体が、酸性条件での使用やAm分離性能の観点から有用であることが示唆された。
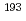 Ir M
Ir M ssbauer spectroscopic parameters of Vaska's complexes and their oxidative adducts
ssbauer spectroscopic parameters of Vaska's complexes and their oxidative adducts金子 政志; 中島 覚*
Inorganic Chemistry, 60(17), p.12740 - 12752, 2021/09
被引用回数:3 パーセンタイル:30.69(Chemistry, Inorganic & Nuclear)高レベル放射性廃液中で観測されている白金族元素と水素との反応の理解に向けて、本研究では、水素と酸化的付加反応を起こす代表的な錯体であるVaska錯体 -[IrCl(CO)(PPh)]とその酸化付加体-[IrCl(YZ)(CO))(PPh)]に着目し、密度汎関数(DFT)計算を用いてIrメスバウアー分光パラメータの報告値と電子状態を相関づけた。DFT計算によるIr錯体の安定構造やIrメスバウアー異性体シフトの再現性を確認した後、Vaska錯体と酸化付加体のメスバウアー分光パラメータの計算を行った。結合解析によって、メスバウアー異性体シフトの傾向が、配位結合における共有結合の強弱と相関づけられることが明らかになった。また、電場勾配(EFG)の解析によって、EFG主軸の符号がYZ=Cl付加体とYZ=H付加体で逆転していることが分かり、状態密度解析から、IrとYZとの配位結合における電子密度分布の違いが、EFG主軸の符号逆転の原因であることが示唆された。
-[IrCl(CO)(PPh)]とその酸化付加体-[IrCl(YZ)(CO))(PPh)]に着目し、密度汎関数(DFT)計算を用いてIrメスバウアー分光パラメータの報告値と電子状態を相関づけた。DFT計算によるIr錯体の安定構造やIrメスバウアー異性体シフトの再現性を確認した後、Vaska錯体と酸化付加体のメスバウアー分光パラメータの計算を行った。結合解析によって、メスバウアー異性体シフトの傾向が、配位結合における共有結合の強弱と相関づけられることが明らかになった。また、電場勾配(EFG)の解析によって、EFG主軸の符号がYZ=Cl付加体とYZ=H付加体で逆転していることが分かり、状態密度解析から、IrとYZとの配位結合における電子密度分布の違いが、EFG主軸の符号逆転の原因であることが示唆された。
深澤 優人*; 金子 政志; 中島 覚*
Journal of Radioanalytical and Nuclear Chemistry, 329(1), p.77 - 84, 2021/07
被引用回数:1 パーセンタイル:15.09(Chemistry, Analytical)マイナーアクチノイドと希土類元素の分離メカニズム解明の一環として、クラウンエーテル型配位子のAm/Eu選択性について密度汎関数計算を用いて研究を行った。まず、異なるリングサイズを持つクラウンエーテル, 12C4, 15C5, 18C6とEuとの錯生成反応を比較した結果、18C6が最も安定に錯体を生成することが分かった。18C6によるAm/Eu選択性を見積った結果、Amに対する選択性が示され、溶媒抽出を用いた既報の研究結果と一致した。18C6の酸素ドナーを窒素及び硫黄ドナーに置換した配位子を用いて、Am/Eu選択性を予測した結果、18C6よりも高いAm選択性を示すことが期待された。化学結合解析の結果、Am-O結合と比較してAm-N及びAm-S結合の強い原子軌道間の相互作用が観測され、その高い共有結合性がクラウンエーテル型配位子によるAm/Eu選択性の起源であることが示唆された。
/Eu selectivity with diethylenetriaminepentaacetic acid and its bisamide chelates.金子 政志; 佐々木 祐二; 松宮 正彦*; 中瀬 正彦*; 竹下 健二*
Journal of Nuclear Science and Technology, 58(5), p.515 - 526, 2021/05
被引用回数:3 パーセンタイル:34.17(Nuclear Science & Technology)マイナーアクチノイドの分離変換技術開発で重要であるAmとEuの金属イオン選択性を理解することを目的として、密度汎関数計算をジエチレントリアミン五酢酸(DTPA)及びそのビスアミド(DTPABA)キレート配位子との金属錯体の分子構造及び錯生成反応のモデル化に適用した。構造最適化計算により得られたDTPA及びDTPABAの錯体構造は、既に報告されている単結晶構造を再現した。錯生成反応におけるギブズエネルギー解析の結果、どちらのキレート配位子ともEuイオンよりAmイオンと安定な錯体を生成することが示され、実験結果のAm/Eu選択性と一致した。金属イオンと配位子との化学結合解析の結果、Amの5f軌道とDTPA及びDTPABAの窒素ドナー原子との強い共有結合が、高いAm選択性が発現した一因であることが示唆された。
complexes of mixed N,O-donor ligands demonstrated on a 10-fold [Eu(TPAMEN)] chelate complexSchnaars, K.; 金子 政志; 藤澤 清史*
Inorganic Chemistry, 60(4), p.2477 - 2491, 2021/02
被引用回数:6 パーセンタイル:56.62(Chemistry, Inorganic & Nuclear)高レベル放射性廃液の有害度低減のために、マイナーアクチノイドに対して高選択的な試薬が必要とされている。混合N,Oドナー配位子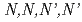 -テトラキス[(6-カルボキシピリジン-2-イル)mメチル]エチレンジアミン(H
-テトラキス[(6-カルボキシピリジン-2-イル)mメチル]エチレンジアミン(H TPAEN)は、Am/Eu分離におけるマスキング剤として良い性能を示すことが知られている。本研究では、Oドナーの塩基性を変えることでM-N
TPAEN)は、Am/Eu分離におけるマスキング剤として良い性能を示すことが知られている。本研究では、Oドナーの塩基性を変えることでM-N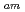 相互作用を操作することが可能なを調査するため、負電荷を帯びたTPAEN
相互作用を操作することが可能なを調査するため、負電荷を帯びたTPAEN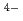 のカルボン酸を中性のアミド基に置換した新規配位子-テトラキス[(6-
のカルボン酸を中性のアミド基に置換した新規配位子-テトラキス[(6-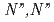 -ジエチルカルバモイルピリジン-2-イル)メチル]エチレンジアミン(TPAMEN)を導入した。TPAMENとEu(OTf)及びEu(NO)6水和物と結晶化することで、正電荷を帯びた[Eu(TPAMEN)]錯体を生成した。M-O/N結合距離を報告されている[Eu(TPAEN)]
-ジエチルカルバモイルピリジン-2-イル)メチル]エチレンジアミン(TPAMEN)を導入した。TPAMENとEu(OTf)及びEu(NO)6水和物と結晶化することで、正電荷を帯びた[Eu(TPAMEN)]錯体を生成した。M-O/N結合距離を報告されている[Eu(TPAEN)] 錯体と比較し、DFT計算の援用により電子密度の違いについて議論した。さらに、[M(TPAEN)]及び[M(TPAMEN)] (M = Eu, Am)の錯生成エネルギーの差の予測によって、TPAMEN配位子のAm/Eu分離に向けた潜在的な性能についての知見を得た。
錯体と比較し、DFT計算の援用により電子密度の違いについて議論した。さらに、[M(TPAEN)]及び[M(TPAMEN)] (M = Eu, Am)の錯生成エネルギーの差の予測によって、TPAMEN配位子のAm/Eu分離に向けた潜在的な性能についての知見を得た。
O)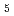 ] with nitrate ions by using density functional theory calculation
] with nitrate ions by using density functional theory calculation加藤 茜*; 金子 政志; 中島 覚*
RSC Advances (Internet), 10(41), p.24434 - 24443, 2020/06
被引用回数:6 パーセンタイル:30.44(Chemistry, Multidisciplinary)高レベル放射性廃液中のルテニウム化学種の安定性を予測することを目的として、硝酸溶液中のルテニウムニトロシル錯体の錯生成反応を密度汎関数法(DFT)を用いて調査した。DFT計算によって得られた[Ru(NO)(NO)(HO)]の最適化構造を既報の実験値と比較した結果、Ru-配位子結合距離やIR振動数を再現することが分かった。幾何異性体間のギブズエネルギーの比較した結果、硝酸イオンの錯生成反応は、Ru-NO軸に対してエクアトリアル平面に配位することによって進行することが明らかになった。また、逐次錯体生成反応におけるギブズエネルギー差を見積ったところ、Ru錯体種と置換する配位子との会合エネルギーを考慮することによって、6M硝酸中のRu錯体種のフラクションを再現することに成功した。Ru-配位子との配位結合の解析の結果、Ru錯体種の安定性は、トランス影響に起因する電子密度の違いによって説明できることが示唆された。本研究は、硝酸中における白金族元素の詳細な錯生成反応のモデル化に寄与することが期待される。
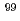 Ru Mssbauer parameters of ruthenium-nitrosyl complexes
Ru Mssbauer parameters of ruthenium-nitrosyl complexes金子 政志; 加藤 茜*; 中島 覚*; 北辻 章浩
Inorganic Chemistry, 58(20), p.14024 - 14033, 2019/10
被引用回数:12 パーセンタイル:62.43(Chemistry, Inorganic & Nuclear)高レベル放射性廃液中に存在することが知られているニトロシルルテニウム錯体について、密度汎関数計算を用いて、Ruメスバウアー分光パラメータ(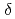 及び
及び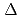
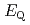 )と配位子場分裂(
)と配位子場分裂(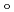 )を相関づけた。[Ru(NO)L] (L = Br, Cl, NH, CN)の構造は、全て報告されている単結晶構造に基づいて作成した。異なるスピン状態で計算した構造とエネルギーを比較した結果、一重項状態[Ru(II)(NO
)を相関づけた。[Ru(NO)L] (L = Br, Cl, NH, CN)の構造は、全て報告されている単結晶構造に基づいて作成した。異なるスピン状態で計算した構造とエネルギーを比較した結果、一重項状態[Ru(II)(NO )L]が最も安定であることが分かった。及びの計算値は、報告されている実験値をよく再現し、L = Br, Cl, NH, CNの順で増加した。さらに、C
)L]が最も安定であることが分かった。及びの計算値は、報告されている実験値をよく再現し、L = Br, Cl, NH, CNの順で増加した。さらに、C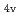 対称性を仮定したを見積った結果、同じ順で増加し、分光化学系列と一致することが分かった。これは、配位子の
対称性を仮定したを見積った結果、同じ順で増加し、分光化学系列と一致することが分かった。これは、配位子の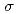 ドナー性及び
ドナー性及び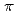 アクセプター性の増加が、結果としてメスバウアー分光パラメータの増加に起因することを示唆している。
アクセプター性の増加が、結果としてメスバウアー分光パラメータの増加に起因することを示唆している。
ssbauer isomer shifts using second-order Douglas-Kroll-Hess Hamiltonian金子 政志; 渡邉 雅之; 宮下 直*; 中島 覚*
Hyperfine Interactions, 239(1), p.20_1 - 20_10, 2018/12
被引用回数:4 パーセンタイル:84.42(Physics, Atomic, Molecular & Chemical)fブロック化合物に対する密度汎関数計算の精度向上を目指し、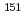 Eu,
Eu, 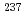 Npメスバウアー異性体シフトを指標として、二次Douglas-Kroll-Hess(DKH2)ハミルトニアンを用いて相対論密度汎関数法のベンチマーク研究を行った。純粋な密度汎関数法による電子交換相互作用とHartree-Fockによる厳密な電子交換相互作用の混合パラメータを変えて、メスバウアー異性体シフトの実験値に対する平均二乗誤差を比較した。その結果、Eu, Npメスバウアー異性体シフトに対して、厳密な交換相互作用の割合が、それぞれ30, 60%のときに、平均二乗誤差が最小になることが明らかになった。
Npメスバウアー異性体シフトを指標として、二次Douglas-Kroll-Hess(DKH2)ハミルトニアンを用いて相対論密度汎関数法のベンチマーク研究を行った。純粋な密度汎関数法による電子交換相互作用とHartree-Fockによる厳密な電子交換相互作用の混合パラメータを変えて、メスバウアー異性体シフトの実験値に対する平均二乗誤差を比較した。その結果、Eu, Npメスバウアー異性体シフトに対して、厳密な交換相互作用の割合が、それぞれ30, 60%のときに、平均二乗誤差が最小になることが明らかになった。
中島 覚*; 金子 政志; 吉浪 啓介*; 岩井 咲樹*; 土手 遥*
Hyperfine Interactions, 239(1), p.39_1 - 39_15, 2018/12
被引用回数:2 パーセンタイル:65.58(Physics, Atomic, Molecular & Chemical)本研究は、鉄二価集積型錯体のスピンクロスオーバー(SCO)のオン・オフ現象と転移挙動の制御について議論する。ビスピリジル型配位子で架橋した鉄二価集積型錯体のSCO現象が起こるか否かは、鉄原子周りの局所構造によって決定する。つまり、鉄原子を通して向き合っているピリジンがプロペラ型で配位しているときにSCO現象は起きるが、平行型または歪んだプロペラ型の場合起きない。これは、プロペラ型の場合、高スピン状態から低スピン状態への構造変化において立体障害がより小さく、鉄とピリジンがお互いに近づくことができることに起因する。さらに、局所構造を制御し、転移温度を変化するための試みとして、ビルディングブロックにメチル基やシステムを導入した配位子の設計や、アニオン配位子の希釈効果についても議論する。
金子 政志; 鈴木 英哉; 松村 達郎
Inorganic Chemistry, 57(23), p.14513 - 14523, 2018/12
被引用回数:20 パーセンタイル:77.49(Chemistry, Inorganic & Nuclear)マイナーアクチノイドの分離変換技術開発の一環として、アメリシウム(Am)とキュリウム(Cm)の分離が課題となっている。本研究では、AmとCmの分離メカニズム解明を目的として、異なる二つのジアミド型配位子であるジグリコールアミド(DGA)とアルキルジアミドアミン(ADAAM)によるAm/Cm選択性の違いを、密度汎関数計算を用いて解析した。モデル錯体として[M(DGA)]と[M(ADAAM)(NO)(HO)]の分子構造探索、錯生成反応ギブズエネルギー計算を行った結果、DGA配位子のCm選択性とADAAM配位子のAm選択性を再現することに成功した。さらに、Am/CmとDGA/ADAAM配位子の化学結合解析を行った結果、結合解離エネルギーの差がAm/Cm選択性の違いに影響を及ぼしており、f軌道電子の共有結合性の違いがAmとCmの分離メカニズムの一因であることが示唆された。
木村 太己*; 金子 政志; 渡邉 雅之; 宮下 直*; 中島 覚*
Dalton Transactions (Internet), 47(42), p.14924 - 14931, 2018/11
被引用回数:9 パーセンタイル:48.5(Chemistry, Inorganic & Nuclear)マイナーアクチノイド(MA)と希土類の分離メカニズム解明を目的として、密度汎関数計算を用いたEu(III)もしくはAm(III)とニクトゲンドナー(X)配位子(CH)X-CH-CH-X(CH) (X=窒素,リン,ヒ素,アンチモン)との錯体に対する計算化学解析を試みた。配位子との錯体生成ギブズエネルギーを解析した結果、リンドナー配位子がEu(III)と比較してAm(III)と安定に錯体を生成することが示唆された。金属(Eu(III)もしくはAm(III))とニクトゲンドナーとの化学結合を解析した結果、Am(III)とリンとの共有結合性が他のニクトゲンに比べて高く、リンドナー配位子が高いAm(III)選択性を有することが示唆された。
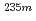 U oxide and fluoride
U oxide and fluoride重河 優大*; 笠松 良崇*; 安田 勇輝*; 金子 政志; 渡邉 雅之; 篠原 厚*
Physical Review C, 98(1), p.014306_1 - 014306_5, 2018/07
被引用回数:4 パーセンタイル:35.62(Physics, Nuclear)Uの半減期は化学的環境に依存して変化することが報告されており、本研究では、初めてUの半減期と内部転換電子エネルギー分光を同一化学的環境下で測定することに成功した。Uの酸化物とフッ化物の試料について測定を行った結果、酸化物に比べフッ化物の半減期は短くなることが観測された。密度汎関数法を用いて内部転換電子エネルギースペクトルのピークを帰属した結果、価電子の化学結合特性の違いが半減期の変化に影響を与えていることが示唆された。
金子 政志; 渡邉 雅之
Journal of Radioanalytical and Nuclear Chemistry, 316(3), p.1129 - 1137, 2018/06
被引用回数:17 パーセンタイル:85.89(Chemistry, Analytical)マイナーアクチノイドとランタノイドの分離メカニズムを理解することを目的として、カルコゲンをドナーとして持つ配位子N(EPMe) (E = O, S, Se, Te)によるAm(III)イオンとEu(III)イオンの選択性を密度汎関数法によって予測した。単結晶構造に基づいて錯体をモデル化し、錯生成による金属イオンの安定化エネルギーを見積もった。その結果、OドナーはAm(III)イオンよりもEu(III)イオンと選択的に錯生成し、S, Se, TeドナーはEu(III)イオンよりもAm(III)イオンと選択的に錯生成する結果となった。この傾向は、HSAB則におけるソフト酸の分類と一致した。金属とカルコゲンの化学結合解析の結果、Am(III)イオンのf軌道との共有結合性が高いドナーほど、Am(III)イオンに対する選択性が高くなることを示唆した。
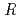 / values by benchmark study of the Mssbauer Isomer shifts for Ru, Os complexes using relativistic DFT calculations
/ values by benchmark study of the Mssbauer Isomer shifts for Ru, Os complexes using relativistic DFT calculations金子 政志; 安原 大樹*; 宮下 直*; 中島 覚*
Hyperfine Interactions, 238(1), p.36_1 - 36_9, 2017/11
被引用回数:2 パーセンタイル:77.36(Physics, Atomic, Molecular & Chemical)Ru, Os錯体の結合状態に対する密度汎関数計算の妥当性を評価することを目的として、Ru, 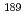 Osのメスバウアー異性体シフト実験値を用いて理論計算手法のベンチマークを行った。結果、Ru錯体およびOs錯体の原子核位置での電子密度の計算値はメスバウアー異性体シフト実験値とよく相関した。
Osのメスバウアー異性体シフト実験値を用いて理論計算手法のベンチマークを行った。結果、Ru錯体およびOs錯体の原子核位置での電子密度の計算値はメスバウアー異性体シフト実験値とよく相関した。
ssbauer isomer shifts with density functional calculations金子 政志; 渡邉 雅之; 宮下 直*; 中島 覚*
Radioisotopes, 66(8), p.289 - 300, 2017/08
Eu錯体の配位結合におけるf電子の役割を理解することを目的として、相対論密度汎関数計算をEu(III)錯体に適用した。既報のEuメスバウアー異性体シフト実験値とEu原子核位置での電子密度計算値の線形性を比較することによって、B2PLYP理論がメスバウアー異性体シフトを最もよく再現することが分かった。また、分子軌道に基づく電子密度の解析によって、d及びf電子が配位結合に大きく関与していることを明らかにした。
金子 政志
放射化学, (35), p.36 - 39, 2017/03
2016年日本放射化学会賞・奨励賞受賞者の受賞対象研究を紹介した解説記事である。メスバウアー分光パラメータと密度汎関数計算を用いて、鉄錯体のスピン相転移現象やランタノイド・アクチノイドの分離メカニズムを明らかにした研究成果について解説を行った。
金子 政志; 渡邉 雅之; 宮下 直*; 中島 覚*
Journal of Nuclear and Radiochemical Sciences (Internet), 17, p.9 - 15, 2017/03
金属イオンの原子価軌道の結合特性の観点から、密度汎関数計算をOドナーであるホスフィン酸とSドナーであるジチオホスフィン酸によるEu(III), Am(III)イオンの錯形成反応に適用した。Sドナー錯体として二つ、Oドナー錯体として四つの幾何異性体の構造最適化を行い、[M(HO)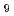 ]に対する錯形成による安定化エネルギーを見積もった。その結果、Oドナー配位子はEu(III)イオンに選択的に配位し、Sドナー配位子はAm(III)イオンに選択的に配位し、実験のAm/Eu選択性を再現した。d,f軌道電子の結合性に着目すると、d軌道電子の結合性に対する寄与はEu, Am錯体のどちらにおいても結合的な特性を持ち、同じ寄与を示した。一方、f軌道電子の寄与は、Eu, Am錯体間で異なり、Sドナー錯体の場合、Euの4f電子は非結合的、Amの5f電子は結合的に振舞うのに対し、Oドナー錯体では、Euの4f電子は結合的、Amの5f電子は反結合的に振舞うことが分かった。この結果から、d軌道電子の結合性は、Eu, Am錯体構造の類似性に、また、f軌道電子の結合性は、Eu, Amイオンの選択性の相違性に起因することが示唆された。
]に対する錯形成による安定化エネルギーを見積もった。その結果、Oドナー配位子はEu(III)イオンに選択的に配位し、Sドナー配位子はAm(III)イオンに選択的に配位し、実験のAm/Eu選択性を再現した。d,f軌道電子の結合性に着目すると、d軌道電子の結合性に対する寄与はEu, Am錯体のどちらにおいても結合的な特性を持ち、同じ寄与を示した。一方、f軌道電子の寄与は、Eu, Am錯体間で異なり、Sドナー錯体の場合、Euの4f電子は非結合的、Amの5f電子は結合的に振舞うのに対し、Oドナー錯体では、Euの4f電子は結合的、Amの5f電子は反結合的に振舞うことが分かった。この結果から、d軌道電子の結合性は、Eu, Am錯体構造の類似性に、また、f軌道電子の結合性は、Eu, Amイオンの選択性の相違性に起因することが示唆された。

