


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
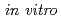 evaluation of F(
evaluation of F(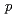 -
- I)KCCYSL for targeting HER2
I)KCCYSL for targeting HER2佐々木 一郎; 渡辺 茂樹; 大島 康宏; 須郷 由美; 山田 圭一*; 花岡 宏史*; 石岡 典子
Peptide Science 2015, p.243 - 246, 2016/03
Radioisotope labeled peptides with high affinity to receptors overexpressing on the surface of tumor cells are promising for applications in nuclear medicine such as diagnostic radiography and radiotherapy. Radiohalogens such as I and  At are useful for clinical imaging and therapeutic applications, and it can be introduced at the
At are useful for clinical imaging and therapeutic applications, and it can be introduced at the 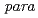 position of phenylalanine residue via electrophilic destannylation. KCCYSL (Lys
position of phenylalanine residue via electrophilic destannylation. KCCYSL (Lys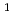 -Cys
-Cys -Cys
-Cys -Tyr
-Tyr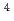 -Ser
-Ser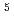 -Leu
-Leu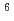 ) is a hexapeptide containing disulfide bond. Previous study revealed that KCCYSL has potential as tumor imaging and therapeutic agent targeting tumor cells overexpressing the human epidermal growth factor receptor type 2 (HER2). In this study, we report synthesis and evaluation of radiohalogenated KCCYSL derivatives. Precursor peptides, Boc-F(-SnBu
) is a hexapeptide containing disulfide bond. Previous study revealed that KCCYSL has potential as tumor imaging and therapeutic agent targeting tumor cells overexpressing the human epidermal growth factor receptor type 2 (HER2). In this study, we report synthesis and evaluation of radiohalogenated KCCYSL derivatives. Precursor peptides, Boc-F(-SnBu )K(Boc)C(Trt)C(Trt)Y(
)K(Boc)C(Trt)C(Trt)Y(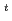 Bu)S(Bu)L-OH and Boc-F(-SnBu)GS(Bu)GK(Boc)C(Trt)C(Trt)Y(Bu)S(Bu)L-OH, were synthesized by the Fmoc solid phase peptide synthesis. Then, precursor peptides were radioiodinated via electrophilic destannylation, and they were deprotected to obtain F(-I)KCCYSL and F(-I)GSGKCCYSL in radiochemical yield 15% and 17%, respectively.
Bu)S(Bu)L-OH and Boc-F(-SnBu)GS(Bu)GK(Boc)C(Trt)C(Trt)Y(Bu)S(Bu)L-OH, were synthesized by the Fmoc solid phase peptide synthesis. Then, precursor peptides were radioiodinated via electrophilic destannylation, and they were deprotected to obtain F(-I)KCCYSL and F(-I)GSGKCCYSL in radiochemical yield 15% and 17%, respectively. 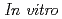 assays of the radioiodinated peptides for HER2 and stability in serum are being undertaken.
assays of the radioiodinated peptides for HER2 and stability in serum are being undertaken.
佐々木 一郎; 花岡 宏史*; 山田 圭一*; 渡辺 茂樹; 須郷 由美; 大島 康宏; 鈴木 博元; 石岡 典子
Peptide Science 2014, p.257 - 260, 2015/03
We have sought to establish drug discovery system using radioisotope (RI) labeled peptides which have high affinity to target proteins overexpressed in cancers. Of the target proteins, we chose the human epidermal growth factor receptor type 2 (HER2), a membrane protein overexpressed in various cancers to evaluate the drug discovery system. Three series of random hexapeptide libraries introduced a radioiodinated D-tyrosine (y(3-I)) to 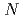 -terminal were designed and binding assay with HER2-expressed cell lines were conducted in this study. First, we synthesized a series of random hexapeptide libraries with fixed amino acid sequence at 1 and 2 positions, y(3-I)XXXXXX. Non-radioactive random peptide libraries, yXXXXXX, were prepared by Fmoc-SPPS with an automatic peptide synthesizer. Radioiodinated y(3-I)XXXXXX were subsequently synthesized in 30-50% radiochemical yield. Binding assay using HER2-overexpressed cell line showed that high affinity (38-50% dose, n=6) was obtained with yIIXXXX, while other random peptide libraries were yielded low affinity (approximately 1% dose), which indicated that the system using RI labeled random peptide libraries have potential to discover peptide drug for cancer therapy. Preparation of other random hexapeptide libraries are being undertaken.
-terminal were designed and binding assay with HER2-expressed cell lines were conducted in this study. First, we synthesized a series of random hexapeptide libraries with fixed amino acid sequence at 1 and 2 positions, y(3-I)XXXXXX. Non-radioactive random peptide libraries, yXXXXXX, were prepared by Fmoc-SPPS with an automatic peptide synthesizer. Radioiodinated y(3-I)XXXXXX were subsequently synthesized in 30-50% radiochemical yield. Binding assay using HER2-overexpressed cell line showed that high affinity (38-50% dose, n=6) was obtained with yIIXXXX, while other random peptide libraries were yielded low affinity (approximately 1% dose), which indicated that the system using RI labeled random peptide libraries have potential to discover peptide drug for cancer therapy. Preparation of other random hexapeptide libraries are being undertaken.
佐々木 一郎; 山田 圭一*; 渡辺 茂樹; 花岡 宏史*; 須郷 由美; 奥 浩之*; 石岡 典子
JAEA-Review 2013-059, JAEA Takasaki Annual Report 2012, P. 96, 2014/03
Radioisotope (RI)-labeled peptide with high affinity to receptors overexpressing on the surface of tumor cells are promising for applications in nuclear medicine such as diagnostic radiography and radiotherapy. MARSGL (H-Met-Ala-Arg-Ser-Gly-Leu-OH), a linear peptide consisting of six amino acids, has high affinity to HER2/neu receptor overexpressing in various cancer cells. Radiohalogens (radionuclides) such as radioiodine and radiobromine are versatile for clinical imaging and therapeutic applications. Thus, radiohalogenated MARSGL may have potential for clinical applications to HER2 overexpressing tumors. In this study, in order to achieve the labeling of radiohalogen for MARSGL, we design a radioiodinated peptide, F(-I)MARSGL, in which phenylalanine labeled with I (t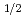 = 8.0 d) at the position of the aromatic ring is introduced into the N-terminal of MARSGL and report a new technique to prepare radiohalogen of peptide via electrophilic destanylation.
= 8.0 d) at the position of the aromatic ring is introduced into the N-terminal of MARSGL and report a new technique to prepare radiohalogen of peptide via electrophilic destanylation.
佐々木 一郎; 山田 圭一*; 渡邉 茂樹; 花岡 宏史*; 須郷 由美; 奥 浩之*; 石岡 典子
Peptide Science 2013, p.157 - 160, 2013/03
Radioisotope (RI)-labeled peptides which have the high affinity to receptors on surface of the tumor cell are promising for diagnostic radiography (PET and SPECT) and radiotherapy. MARSGL (H-Met-Ala-Arg-Ser-Gly-Leu-OH), a linear hexapeptide consisting of six amino acids, has the high affinity to HER2/neu receptor overexpressing in various cancer cells. Thus, radiolabeled MARSGL has potential for the above mentioned purposes. Radiohalogens have various useful radionuclides, which could be introduced to aromatic compounds via tin-halogen exchange reaction. In this study, stannylated peptide Boc-F(-SnBu)MAR(Pbf)S(Bu)GL-OH was synthesized to prepare radiohalogen (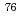 Br or I)-labeled FMARSGL derivatives. First, Boc-Phe(-SnBu)-OH was prepared from Boc-Phe(-I)-OMe via Pd(0)-catalyzed coupling reaction with (BuSn)
Br or I)-labeled FMARSGL derivatives. First, Boc-Phe(-SnBu)-OH was prepared from Boc-Phe(-I)-OMe via Pd(0)-catalyzed coupling reaction with (BuSn) and was successfully introduced into
and was successfully introduced into 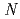 -terminal of H-MARSGL-Trt(2-Cl) resin synthesized from H-Leu-Trt(2-Cl) resin by Fmoc-SPPS. After cleavage reaction of Boc-F(-SnBu)MAR(Pbf)S(Bu)GL-Trt(2-Cl) resin with 25% 1,1,1,3,3,3-hexafluoro-2-propanol in CHCl, Boc-F(-SnBu)MAR(Pbf)S(Bu)GL-OH was obtained and identified by using ESI-MS and NMR. Synthesis of I-labeled FMARSGL is due to report in this presentation.
-terminal of H-MARSGL-Trt(2-Cl) resin synthesized from H-Leu-Trt(2-Cl) resin by Fmoc-SPPS. After cleavage reaction of Boc-F(-SnBu)MAR(Pbf)S(Bu)GL-Trt(2-Cl) resin with 25% 1,1,1,3,3,3-hexafluoro-2-propanol in CHCl, Boc-F(-SnBu)MAR(Pbf)S(Bu)GL-OH was obtained and identified by using ESI-MS and NMR. Synthesis of I-labeled FMARSGL is due to report in this presentation.
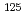 I]-sansalvamide A derivative
I]-sansalvamide A derivative渡邉 茂樹; 山田 圭一*; 津久井 匠隆*; 花岡 宏史*; 大島 康宏; 山口 藍子*; 奥 浩之*; 石岡 典子
JAEA-Review 2012-046, JAEA Takasaki Annual Report 2011, P. 88, 2013/01
Sansalvamide A (SA), a penta cyclic peptide isolated marine fungus, is a lead compound of anti-cancer reagent because the peptide has cytotoxicity against various cancer cell lines. Halogenated SA derivatives (SA-X, X = Cl, Br, I) was prepared and remarkable cytotocity against malignant human breast cancer. In this study, a radiohalogenated SA derivative [I]SA-I was prepared to conduct in vivo evaluation of SA derivatives. Synthetic scheme of [I]SA-I are as follows: an iodinated linear peptide, Boc-F(p-I)LLVL-OMe, was prepared by the conventional solid phase peptide synthesis. After preparation of stannylated peptide, Boc-F(p-SnBu)LLVL-OMe, I was labeled with electrophilic destannylation in the presence of oxidizing reagent. After deprotection of N- and C-termius, [I]SA-I was obtained successfully by macrocyclization in liquid phase. Overall labeled yield was 7%. To our best knowledge, this report is the first on the synthesis of radiolabeled SA derivative. In vivo evaluation of the SA derivative using [I]SA-I is being undertaken.
Br-labeled amino acid derivative for PET imaging of tumor花岡 宏史*; 渡邉 茂樹; 富永 英之*; 大島 康宏; 渡辺 智; 山田 圭一*; 飯田 靖彦*; 石岡 典子; 遠藤 啓吾*
JAEA-Review 2012-046, JAEA Takasaki Annual Report 2011, P. 89, 2013/01
近年、がんに対する特異性が高いPET薬剤として、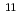 Cや
Cや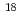 Fで標識したアミノ酸誘導体が開発され、臨床応用されるようになってきた。しかしながらCやFは半減期が非常に短いため、それぞれの病院で製造・合成する必要があり、限られた施設でしか使えないのが現状である。一方、Brは、半減期が16.1時間とポジトロン放出核種としては比較的長く、またハロゲン核種であるため母体化合物との結合にキレート剤等が必要ないことから、アミノ酸のような低分子化合物に対しても応用可能である。そこで本研究では、広く臨床使用することが可能な、新規がん診断用PETイメージング薬剤としてBr標識アミノ酸誘導体の開発を計画した。基礎検討には半減期が長い放射性臭素である
Fで標識したアミノ酸誘導体が開発され、臨床応用されるようになってきた。しかしながらCやFは半減期が非常に短いため、それぞれの病院で製造・合成する必要があり、限られた施設でしか使えないのが現状である。一方、Brは、半減期が16.1時間とポジトロン放出核種としては比較的長く、またハロゲン核種であるため母体化合物との結合にキレート剤等が必要ないことから、アミノ酸のような低分子化合物に対しても応用可能である。そこで本研究では、広く臨床使用することが可能な、新規がん診断用PETイメージング薬剤としてBr標識アミノ酸誘導体の開発を計画した。基礎検討には半減期が長い放射性臭素である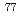 Br(半減期57時間)を用いて行うこととした。Br標識アミノ酸としては、
Br(半減期57時間)を用いて行うこととした。Br標識アミノ酸としては、 メチルフェニルアラニン(-Me-Phe)のパラ位にBrを導入したBr--Me-Pheを設計した。Br--Me-Pheは標識率25-40%で合成することができた。Br--Me-Pheを担癌マウスに投与したところ、腫瘍への高い集積性を示し、投与3時間後の腫瘍対血液比は3.94、腫瘍対筋肉比は3.95であった。Br--Me-Pheを担癌マウスに投与してPET撮像を行ったところ、腫瘍を明瞭に描出することができた。以上の結果から、Br--Me-Pheの新規がんイメージング薬剤としての有用性が示唆された。
メチルフェニルアラニン(-Me-Phe)のパラ位にBrを導入したBr--Me-Pheを設計した。Br--Me-Pheは標識率25-40%で合成することができた。Br--Me-Pheを担癌マウスに投与したところ、腫瘍への高い集積性を示し、投与3時間後の腫瘍対血液比は3.94、腫瘍対筋肉比は3.95であった。Br--Me-Pheを担癌マウスに投与してPET撮像を行ったところ、腫瘍を明瞭に描出することができた。以上の結果から、Br--Me-Pheの新規がんイメージング薬剤としての有用性が示唆された。
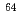 Cu-labeled DOTA-
Cu-labeled DOTA-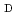 -Phe-Tyr-octreotide (Cu-DOTA-TOC) for imaging somatostatin receptor-expressing tumors
-Phe-Tyr-octreotide (Cu-DOTA-TOC) for imaging somatostatin receptor-expressing tumors花岡 宏史*; 富永 英之*; 山田 圭一*; Paudyal, P.*; 飯田 靖彦*; 渡邉 茂樹; Paudyal, B.*; 樋口 徹也*; 織内 昇*; 遠藤 啓吾*
Annals of Nuclear Medicine, 23(6), p.559 - 567, 2009/08
被引用回数:17 パーセンタイル:46.65(Radiology, Nuclear Medicine & Medical Imaging)In-111 (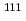 In)-labeled octreotide has been clinically used for imaging somatostatin receptor-positive tumors, and radiolabeled octreotide analogs for positron emission tomography (PET) have been developed. The aim of this study is to produce and fundamentally examine a Cu-labeled octreotide analog, Cu-1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid--Phe-Tyr-octreotide (Cu-DOTA-TOC). Cu-DOTA-TOC can be produced in amounts efficient for clinical study with high radiochemical yield. Cu-DOTA-TOC was stable in vitro, but time-dependent transchelation to protein was observed after injection into mice. In biodistribution studies, the radioactivity of Cu was higher than that of In in all organs except kidney. In tumor-bearing mice, Cu-DOTA-TOC showed a high accumulation in the tumors, and the tumor-to-blood ratio reached as high as 8.81
In)-labeled octreotide has been clinically used for imaging somatostatin receptor-positive tumors, and radiolabeled octreotide analogs for positron emission tomography (PET) have been developed. The aim of this study is to produce and fundamentally examine a Cu-labeled octreotide analog, Cu-1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid--Phe-Tyr-octreotide (Cu-DOTA-TOC). Cu-DOTA-TOC can be produced in amounts efficient for clinical study with high radiochemical yield. Cu-DOTA-TOC was stable in vitro, but time-dependent transchelation to protein was observed after injection into mice. In biodistribution studies, the radioactivity of Cu was higher than that of In in all organs except kidney. In tumor-bearing mice, Cu-DOTA-TOC showed a high accumulation in the tumors, and the tumor-to-blood ratio reached as high as 8.81 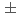 1.17 at 6 h after administration. Cu-DOTA-TOC showed significantly higher accumulation in the tumor than Cu-TETA-OC and Cu-DOTA-OC. PET showed a very clear image of the tumor, which was comparable to that of F-FDG PET and very similar to that of Cu-TETA-OC. Cu-DOTA-TOC clearly imaged a somatostatin receptor-positive tumor and seemed to be a potential PET tracer in the clinical phase.
1.17 at 6 h after administration. Cu-DOTA-TOC showed significantly higher accumulation in the tumor than Cu-TETA-OC and Cu-DOTA-OC. PET showed a very clear image of the tumor, which was comparable to that of F-FDG PET and very similar to that of Cu-TETA-OC. Cu-DOTA-TOC clearly imaged a somatostatin receptor-positive tumor and seemed to be a potential PET tracer in the clinical phase.
 enolase
enolase奥 浩之*; 山田 圭一*; 小林 京子*; 片貝 良一*; Ashfaq, M.*; 花岡 宏史*; 飯田 靖彦*; 遠藤 啓吾*; 長谷川 伸; 前川 康成; et al.
Peptide Science 2008, p.439 - 442, 2009/03
マラリアは、熱帯及び亜熱帯地域における主な死因の一つである。これまでの研究において、エノラーゼ基質結合部位の部分配列に由来する人工ペプチド抗原としてマラリアワクチン抗原の有用性を検証してきた。人工抗原ペプチドは、抗原性ペプチドに5(6)-カルボキシフルオロセインを用いて蛍光ラベルして合成した。合成したペプチドは、乳酸とグリコール酸の共重合体(PLGA)を用いて乳化重合後、ホモジナイズし、ナノ粒子化した。この粒子へ再度0.5%ポリビニルアルコールを加えた後、乳化,ホモジナイズして粒子径0.3から1.5mmのナノ粒子を調製した。蛍光強度からみた生体外での徐放試験において、ペプチド抗原のみから作製したナノ球体を用いた場合、薬剤は、保留日数に対してほぼ0次で急激に放出されるのに比べ、PLGAナノ粒子に調製した試料は、1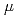 g/7日間で徐放されることがわかった。1.0mg(蛍光入り薬剤4.0g)のナノ粒子を用いたハダカネズミによる生体内試験において、蛍光強度は、12日間かけ次第に減少し、今回調製されたナノ微粒子は持続的に抗原を徐放することがわかった。
g/7日間で徐放されることがわかった。1.0mg(蛍光入り薬剤4.0g)のナノ粒子を用いたハダカネズミによる生体内試験において、蛍光強度は、12日間かけ次第に減少し、今回調製されたナノ微粒子は持続的に抗原を徐放することがわかった。
藤原 治; 大井 圭一*; 山田 和芳*; 加藤 めぐみ*; 小野 有五*; 伊勢 明広*
月刊地球, p.463 - 468, 1997/00
気候・海水準は、地層処分で対象とする長期的将来には現在と大きく変わると考えられる。この為気候・海水順の変化幅や周期性を明らかにし、地下水の涵養量や水質の変化と地質環境の長期安定性に影響を与える情報を整理しておくことが重要である。本研究では、湖沼や内湾等の堆積物を利用して、日本列島周辺の降水量変動などを支配するアジアモンスーンの変動を、過去3.5万年間について調べた。この結果は、国際的にも注目されているアジア地域の気候変動研究に寄与するとともに、過去の気候変動を検出する為の手法の確立にも寄与する。 なお本稿は、月刊地球からの要請で投稿・講演するものである。
山澤 弘実; 永井 晴康; 茅野 政道; 林 隆; 守屋 省三*; 高橋 圭一*; 小島 啓美*; 山田 智久*
JAERI-Data/Code 95-012, 65 Pages, 1995/09
 JAERI-Data-Code-95-012.pdf:2.24MB
JAERI-Data-Code-95-012.pdf:2.24MB
本報告書は、平成5年12月に原研東海研構内の松林を対象に実施した小規模な森林内拡散実験および気象・乱流観測の結果をまとめたものである。拡散実験では、森林内外に設置した線状放出源からの鉛直拡散に着目し、4地点に高さ方向に設置した捕集装置により濃度分布を取得した。得られた拡散データは、図表の形で本報告書に収録した。気象・乱流観測では、超音波風向風速計を森林内外に5ヶ所設置して10Hzのサンプリングで行った。得られたデータは量が膨大で、今後種々の解析で使用されることを考え、統一した形式で光磁気ディスクに収録した。本報告書には、10分平均値及びデータファイルの収録形式のみを記載した。
津久井 匠隆; 渡邉 茂樹; 山田 圭一*; 花岡 宏史*; 奥 浩之*; 松尾 一郎*; 遠藤 啓吾*; 石岡 典子
no journal, ,
生理活性ペプチドの中には、抗がん作用,抗菌作用を示すものやホルモン様作用を示して受容体と結合するものがあり、創薬のリード化合物となり得る。われわれが開発したパラ位に臭素原子が置換された芳香族アミノ酸を構造中に有する環状ペプチドは、インビトロ実験において乳がん細胞の増殖抑制能を有することが明らかになっている。そこで、インビボ実験における体内分布の取得を目指し、開発した環状ペプチドへの放射性臭素の標識に関する基礎検討を実施した。構成アミノ酸である芳香族アミノ酸誘導体の標識条件では、前駆体に対しCATを1等量、NCSでは4等量加え、室温15分間反応させることで、それぞれ標識率70%で反応が進行し、効率よくBr-77を導入できることを確認した。標識前駆体の合成では、Boc-SPPS法により生成した化合物についてESI-MS分析を行った結果、目的物に相当するイオンピークは得られず、トリブチルスズアミノ酸を用いるBoc-SPPS法は環状ペプチド合成に適していないことが明らかとなった。そこで、現在新たな合成法として脱保護に塩基を用いるFmoc-SPPSによる合成について検討を行っている。
津久井 匠隆; 渡邉 茂樹; 花岡 宏史*; 山田 圭一*; 奥 浩之*; 石岡 典子; 遠藤 啓吾*; 松尾 一郎*
no journal, ,
生理活性ペプチドには顕著な抗腫瘍活性を示すものがあり、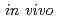 での治療効果に興味が持たれている。これらの生理活性ペプチドにおける構造活性相関等の創薬研究は、さまざまな所で行われている。創薬研究では、薬剤の体内分布や腫瘍への集積能が重要な因子となり得る。演者らは、放射性核種を標識した生理活性ペプチドを用いて体内での薬剤挙動と治療効果を同時に計測できれば薬剤開発の効率化が可能になると考え、その標識化法を検討している。本発表では、Boc-Phe(
での治療効果に興味が持たれている。これらの生理活性ペプチドにおける構造活性相関等の創薬研究は、さまざまな所で行われている。創薬研究では、薬剤の体内分布や腫瘍への集積能が重要な因子となり得る。演者らは、放射性核種を標識した生理活性ペプチドを用いて体内での薬剤挙動と治療効果を同時に計測できれば薬剤開発の効率化が可能になると考え、その標識化法を検討している。本発表では、Boc-Phe(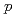 -SnBu)-OMeを前駆体としたBr-77標識アミノ酸の合成に関する基礎的検討について報告する。酸化剤にChloramine T、N-Chlorosuccinimideを用いて室温30分間標識化したところ、その標識率は60
-SnBu)-OMeを前駆体としたBr-77標識アミノ酸の合成に関する基礎的検討について報告する。酸化剤にChloramine T、N-Chlorosuccinimideを用いて室温30分間標識化したところ、その標識率は60 85%であった。現在、Phe(-SnBu)含有ペプチドへの適用も検討しており、併せて報告する予定である。
85%であった。現在、Phe(-SnBu)含有ペプチドへの適用も検討しており、併せて報告する予定である。
Br標識アミノ酸誘導体の開発花岡 宏史*; 渡邉 茂樹; 富永 英之*; 大島 康宏; 津久井 匠隆; 渡辺 智; 山田 圭一*; 飯田 靖彦*; 織内 昇*; 樋口 徹也*; et al.
no journal, ,
CやFで標識したアミノ酸誘導体は、がん診断用のPET薬剤として、臨床使用されているものも少なくない。しかしながらCやFは短半減期であるため、それぞれの病院で製造する必要があり、限られた施設でしか使えないのが現状である。一方でBrは半減期が16.1時間とポジトロン放出核種としては比較的長く、またFと同じハロゲン核種であるため、F標識アミノ酸製剤と同様の分子設計を行うことができるという利点を有する。そこで本研究では、広く臨床使用することが可能な、新規がん診断用PETイメージング薬剤としてBr標識アミノ酸誘導体を開発することを計画した。基礎的検討には半減期が長い放射性臭素であるBr(半減期57時間)を用いて行うこととした。Br標識アミノ酸としては、-メチルフェニルアラニンのパラ位にBrを導入したp-Bromo--methyl-Pheを設計した。Br標識Phe誘導体は標識率2540%で合成することができた。ノーマルマウスにおける体内動態を検討した結果、膵臓に高い集積が認められたことから、本化合物はアミノ酸トランスポーターに認識されていることが示唆された。一方で腎臓に対しても高い集積がみられた。担癌マウスに投与したところ、腫瘍への高い集積性を示し、投与3時間後の腫瘍血液比は3.94、腫瘍筋肉比は3.51となった。以上の結果から、Br標識Phe誘導体は、新規がんイメージング剤として有用である可能性が示された。
津久井 匠隆; 山田 圭一*; 渡邉 茂樹; 花岡 宏史*; 奥 浩之*; 遠藤 啓吾*; 石岡 典子; 松尾 一郎*
no journal, ,
p-(tri-n-butyl-stannyl)-phenylalanine (Phe(p-BuSn)) is an useful building blocks for synthesizing radiolabeled pharmaceutics. To best of our knowledge, there are few studies of the synthesis of Phe(p-BuSn)-containing peptides. In this study, we report solid-phase synthesis of cyclic peptides containing Phe(p-BuSn) residue, cyclo(Phe(p-BuSn)-Leu-MeLeu-Val-Leu) (I). Boc-Phe(p-BuSn)-OH was synthesized from Boc-Phe(p-I)-OMe by treatment with bis(tri-n-butyl)tin and Pd(0) catalyst followed by saponification. The linear peptide containing Phe(p-BuSn) residue was assembled on Kaiser-oxime resin. On-resin macrocyclization of the linear peptide, however, did not afford the cyclic peptide I. Cross coupling of cyclic peptide containing Phe(p-I) residue, cyclo(Phe(p-I)-Leu-MeLeu-Val-Leu) with bis(tri-n-butyl)tin and Pd(0) catalyst gave the desired peptide, which was confirmed by ESI-MS. In this presentation, we will report the Fmoc-SPPS of the peptide I.
I-Sansalvamide Aの合成と評価渡邉 茂樹; 津久井 匠隆; 花岡 宏史*; 山田 圭一*; 大島 康宏; 飯田 靖彦*; 遠藤 啓吾*; 石岡 典子
no journal, ,
抗がん作用や抗菌作用を示す生理活性物質は、創薬におけるリード化合物となる。Sansalvamide A(SA)は、構造中にエステル結合を有する海洋真菌類由来の環状デプシペプチドで、大腸がん細胞に対して高い細胞毒性を示すことから、抗がん剤開発のリード化合物として注目されている。これまでわれわれは、フェニルアラニン側鎖にヨウ素を導入したSA誘導体を開発し、この化合物が悪性度の高い乳がん細胞MD-MBA-231に対して高い細胞障害活性を示すことを明らかにしてきた。そこで、本研究ではI-SAの生体内における動態を解明するために、Iを導入したI-SAを合成し、ノーマルマウスにおける体内分布を明らかにすることとした。
山田 圭一*; 渡邉 茂樹; 大島 康宏; 花岡 宏史*; 津久井 匠隆*; 高野 智香子*; 山口 藍子*; 奥 浩之*; 石岡 典子
no journal, ,
Sansalvamide A (SA) is a marin-derived cyclic depsipeptide that exhibits cytotoxicity against colon cancer cell line. Recently, a series of cyclic pentapeptide analogs of SA in which Hmp ((2S)-2-hydroxy-4-methylpentanoic acid) residue was replaced by amino acids or N-methyl-amino acids were synthesized. We previously reported that the haogenated analog of [MeLeu]SA, namely SA-Br, SA-I, SA-Cl, exhibited potent cytotoxicity against MDA-MB-231. In the present study, we considered to evaluate in vivo biodistribution, stability and antitumor activity of these halogenated SA peptides in tumor-bearing mice. For this purpose, radiohalogen atoms are most suitable for labeling without changing their primary structure and biological activity. Here, we report the synthesis and in vivo biodistribution of I-labeled SA analog (SA-[I]).
I標識環状ペプチドSansalvamide A誘導体の合成と評価渡邉 茂樹; 山田 圭一*; 花岡 宏史*; 大島 康宏; 津久井 匠隆*; 高野 智香子*; 山口 藍子*; 奥 浩之*; 飯田 靖彦*; 遠藤 啓吾*; et al.
no journal, ,
Sansalvamide A(SA)は、抗腫瘍活性を持つ海洋真菌類由来の天然環状ペプチドである。われわれはSAのPhe側鎖芳香環にハロゲンを導入した誘導体SA-X(cyclo(Phe(p-X)-Leu-MeLeu-Val-Leu), X=Cl, Br, I)を合成し、これらが乳がん細胞MDA-MB-231に対してin vitroで細胞毒性を持つことを見いだした。本研究ではin vivo評価を目的としてI標識Sansalvamide A誘導体[I]SA-Iの合成について検討した。その結果、Boc固相合成法により直鎖ペプチドBoc-Phe(p-SnBu)-Leu-MeLeu-Val-Leu-OMeを得た後、スズ-ヨウ素交換反応によるI標識,脱保護、及び、液相での環化を行うことで[I]SA-Iを合成できることを見いだした。
渡邉 茂樹; 西中 一朗; 山田 圭一*; 橋本 和幸; 牧井 宏之; 花岡 宏史*; 石岡 典子
no journal, ,
At-211は内用放射線治療への応用が期待されている線放出核種である。At-211をがん細胞などに過剰発現する受容体に対して高い親和性を持つ生体分子に標識することで、がんの内用放射線治療への応用が可能になる。本発表では内用放射線治療に向けた基礎検討としてアミノ酸に着目し、放射性ハロゲン標識で最も用いられているスズ-アスタチン交換反応を用いたアミノ酸誘導体へのAt-211標識について報告する。
渡邉 茂樹; 山田 圭一*; 花岡 宏史*; 大島 康宏; 高野 智香子*; 津久井 匠隆*; 山口 藍子*; 須郷 由美; 奥 浩之*; 石岡 典子
no journal, ,
がん細胞に対する細胞障害活性を持つ生理活性ペプチドは、抗がん剤開発におけるリード化合物(化学修飾することで活性の向上、毒性の減弱などが期待できる新規化合物)となりうる。中でも環状ペプチドは、ペプチド分解酵素に対する耐性が高く、リジッドな構造を持つことから、生体内において安定で標的細胞に対して選択性が高い抗がん薬剤のリード化合物として期待できる。本発表では、4つのアミノ酸と1つのヒドロキシ酸から構成される海洋真菌類由来の環状ペプチドSansalvamide A(SA)をリード化合物として開発した難治性乳がん細胞に対する細胞障害活性を持つSA-Iの体内動態を明らかにするために、放射性ヨウ素であるI-125(半減期59.4日)を導入した[I]SA-Iの合成、及び、ノーマルマウス及び難治性乳がん移植マウスにおける体内動態の検討について報告する。
Br標識アミノ酸の開発花岡 宏史*; 渡邉 茂樹; 富永 英之*; 大島 康宏; 渡辺 智; 山田 圭一*; 荒野 泰*; 石岡 典子; 遠藤 啓吾*
no journal, ,
近年、がんに対する特異性が高いPET薬剤として、CやFで標識したアミノ酸誘導体が開発され、臨床応用されるようになってきた。しかしながらCやFは半減期が非常に短いため、それぞれの病院で製造・合成する必要があり、限られた施設でしか使えないのが現状である。一方、Brは、半減期が16.1時間とポジトロン放出核種としては比較的長く、またハロゲン核種であるため母体化合物との結合にキレート剤等が必要ないことから、アミノ酸のような低分子化合物に対しても応用可能である。そこで本研究では、広く臨床使用することが可能な、新規がん診断用PETイメージング薬剤としてBr標識アミノ酸誘導体の開発を計画した。基礎検討には半減期が長い放射性臭素であるBr(半減期57時間)を用いて行うこととした。Br標識アミノ酸としては、メチルフェニルアラニン(-Me-Phe)のパラ位にBrを導入したBr--Me-Pheを設計した。Br--Me-Pheは標識率25-40%で合成することができた。Br--Me-Pheを担癌マウスに投与したところ、腫瘍への高い集積性を示し、投与3時間後の腫瘍対血液比は3.94、腫瘍対筋肉比は3.95であった。Br--Me-Pheを担癌マウスに投与し、6時間後にPET撮像を行ったところ、腫瘍を明瞭に描出することができた。以上の結果から、Br--Me-Pheの新規がんイメージング薬剤としての有用性が示唆された。
